Scout: more local observations than total number of samples
I think there's an issue with the count of local observations (archive 2017-05-31)? I'm looking at mitochondrial variants and there are more observations than there are total number of samples.
Below are images from case 20017, variants https://scout.scilifelab.se/cust003/20017/e3b5c7e0ce3f8478ea0bb45decb003a9 and https://scout.scilifelab.se/cust003/20017/26b2b4bf58686330c5f6c94a61acad27
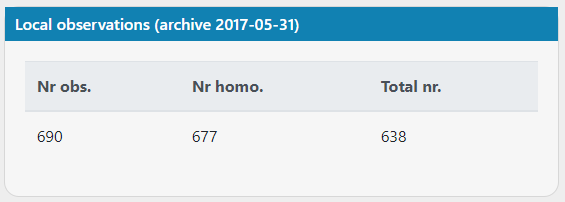
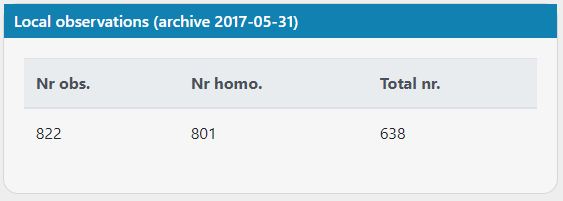
It doesn't affect my interpretation of the variants, but something seems a little off :)
/Ingegerd
 ielvers
ielvers
All 19 comments
Hehe yes it is time to take care of this one. I think that the date and the total nr is hardcoded and not updated in a long time. This information should be parsed from the VCF-header and stored on case level.
 moonso
on 29 Oct 2020
moonso
on 29 Oct 2020
For the same mitochondrial variants, the local observations haven't increased much between the archive and the current database. Are new cases added to the archived local observations for mitochondrial variants?
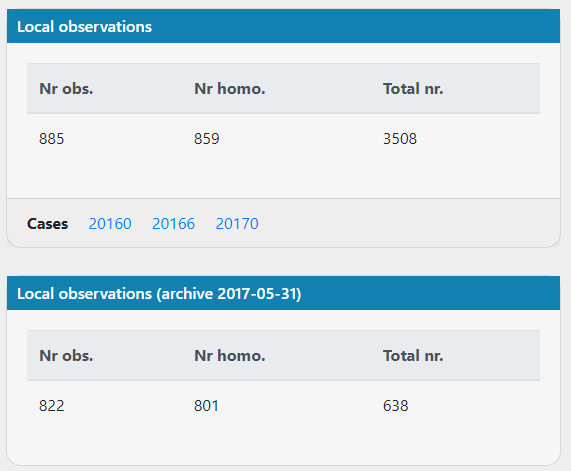
Even for variants that are less abundant, it seems impossible that in the current database with >5x as many cases it's essentially the same number of observations. Picture below from variant https://scout.scilifelab.se/cust003/20017/946b2c1ab17afe5d9a8da513bf08f3d4
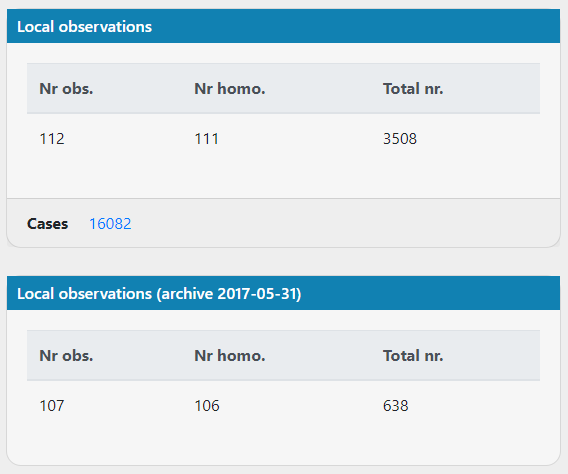
ielvers
on 29 Oct 2020
Quite right @ielvers. It’s this issue: #1559. We are presumably waiting for that date and nr of cases/chromosomes to land in a data file @moonso?
 dnil
on 29 Oct 2020
dnil
on 29 Oct 2020
Are they not already in the VCF header? Otherwise we should open an issue in loqusdb
moonso
on 29 Oct 2020
Well, you are assigned so.. 😉😸
dnil
on 29 Oct 2020
I understand that this is not top priority among all the other things you're working on, but do you think it would be possible to remove the date+total cases or alternatively list both dates+total cases (2017 and 2019) until a permanent solution is in place? As a heads up, since apparently at least everyone in my office had missed this for the past year. :)
ielvers
on 29 Oct 2020
@moonso the dates are actually sort of available from the VCF header, since they are part of the MIP reference file names for the loqus dumps; e.g.
##INFO=<ID=Obs,Number=1,Type=Integer,Description="The number of observations for the variant (from /home/proj/production/rare-disease/references/references_9.0/grch37_loqusdb_snv_indel_-2020-08-21-.vcf.gz)">
The count of chromosomes or individuals is as far as I can tell only present in that dump file? It would not seem impossible to transfer that as part of the annotation. Did we have a MIP issue for this somewhere or should we perhaps make one (@henrikstranneheim)?
dnil
on 5 Nov 2020
Yes let's discuss this together with @henrikstranneheim and @jemten . I think it will be a fairly easy to fix. It's just that we need to coordinate loqusdb, mip and scout
moonso
on 6 Nov 2020
Ping @moonso and @henrikstranneheim! 😸 We'd need to propagate that dump file individual count (and perhaps ideally date) through MIP and CG to Scout. Worst case we could script it, adding a value directly to the db by going to online vcf headers, checking the reference files to collect the metadata. But prospective is our usual mode of operation..
dnil
on 22 Dec 2020
This is what the loqusdb dump file header looks like:
##fileformat=VCFv4.3
##FILTER=<ID=PASS,Description="All filters passed">
##INFO=<ID=Obs,Number=1,Type=Integer,Description="The number of observations for the variant">
##INFO=<ID=Hom,Number=1,Type=Integer,Description="The number of observed homozygotes">
##INFO=<ID=Hem,Number=1,Type=Integer,Description="The number of observed hemizygotes">
##contig=<ID=1,length=249240620>
##contig=<ID=2,length=243189191>
##contig=<ID=3,length=197961645>
##contig=<ID=4,length=191044293>
##contig=<ID=5,length=180905168>
##contig=<ID=6,length=171053709>
##contig=<ID=7,length=159128666>
##contig=<ID=8,length=146303967>
##contig=<ID=9,length=141153153>
##contig=<ID=10,length=135524774>
##contig=<ID=11,length=134946515>
##contig=<ID=12,length=133841873>
##contig=<ID=13,length=115109853>
##contig=<ID=14,length=107289492>
##contig=<ID=15,length=102521387>
##contig=<ID=16,length=90294733>
##contig=<ID=17,length=81195191>
##contig=<ID=18,length=78017152>
##contig=<ID=19,length=59118988>
##contig=<ID=20,length=62965514>
##contig=<ID=21,length=48119891>
##contig=<ID=22,length=51244530>
##contig=<ID=X,length=155260500>
##contig=<ID=Y,length=59034001>
##contig=<ID=MT,length=16567>
##NrCases=3236
##Software=<ID=loqusdb,Version=2.5,Date="2020-08-21 09:02",CommandLineOptions="">
##bcftools_viewVersion=1.10.2+htslib-1.10.2
##bcftools_viewCommand=view --output-type v /home/proj/stage/rare-disease/references/references_9.0/grch37_loqusdb_snv_indel_-2020-08-21-.vcf.gz; Date=Mon Aug 24 19:03:16 2020
##INFO=<ID=OLD_MULTIALLELIC,Number=1,Type=String,Description="Original chr:pos:ref:alt encoding">
##INFO=<ID=OLD_VARIANT,Number=.,Type=String,Description="Original chr:pos:ref:alt encoding">
#CHROM POS ID REF ALT QUAL FILTER INFO
 henrikstranneheim
on 22 Dec 2020
henrikstranneheim
on 22 Dec 2020
I just do not really have a clear idea of where MIP would parse and add annotation info on NrCases and the dump file version
henrikstranneheim
on 22 Dec 2020
I guess we can make a sub to parse the info - put it in a yaml file like loqusdb_reference_info.yaml:
NrCases: <NUMBER_OF_CASES>
Date: <DUMP_DATE>
and add the loqusdb_reference_info.yaml to HK with a proper tag to be picked up by Scout.
or we have to add the NrCases and a date as header to the VCF and scout will have to parse the indata file
henrikstranneheim
on 22 Dec 2020
Either works for me! Slight preference for the latter to make it easier for others to mimic, but really, artistic license on that one!
dnil
on 22 Dec 2020
The case VCF header that is, otw it is probably better with a yaml. Having scout go search for a reference file on the fs, parse it and add stuff to db is a little odd. Id rather script it / cg it then.
dnil
on 22 Dec 2020
Yes, if not case VCF, then one of the yamls in hk, preferentially one that cg already parses, so we could lift the values over to the scout load config in cg?
dnil
on 22 Dec 2020
Yes, the VCF variants file header - not the loqus dump vcf
henrikstranneheim
on 22 Dec 2020
Added NrCases and loqusdb software line, which contains the date of the dump and loqusDB versionto variant VCF header here: https://github.com/Clinical-Genomics/MIP/pull/1763
henrikstranneheim
on 22 Dec 2020
This should now be available with MIP 10 in Sthlm
henrikstranneheim
on 16 Jun 2021
Then we have to parse it out of the VCF!
 northwestwitch
on 16 Jun 2021
northwestwitch
on 16 Jun 2021
Related issues
 hassanfa
·
3Comments
northwestwitch
·
5Comments
hassanfa
·
3Comments
northwestwitch
·
5Comments
 heronikdin
·
4Comments
heronikdin
·
4Comments
 mhkc
·
3Comments
mhkc
·
3Comments
 andreaswallberg
·
4Comments
andreaswallberg
·
4Comments
Most helpful comment
This should now be available with MIP 10 in Sthlm