Scout: Getting pretend-data into Scout
Dear developers,
I am new to Scout and have some questions.
We are developing a lab for a class where we aim to use Scout to trace a Mendelian disease in a pedigree. In the absence of real clinical data we are thinking of taking some chromosomes from the 1000 genomes project as founders and simulate offspring and matings. After that we will seed the resulting individuals with the particular mutation.
This all requires quite some work, and will result in some VCF, PED and MAP files with variants and some basic relationships between individuals.
My first question is, how is this data normally loaded into Scout? Is it normally side-loaded from command-line, or uploaded by a user via the web?
The examples available in the scout repo have a lot of metadata spread out across various yaml and variant files. Is there a way to easily generate the needed metadata or do the files need to be hand-crafted? What is the smallest viable set of metadata that can be used to load variation from VCF, a pedigree and encoding for whether someone is affected or not (or carriers/non-carriers)?
Lastly, as long as it is specified (somewhere?), does it matter whether we work with variants scored against the Ch37 or Ch38 human genome assemblies? The examples seem to use Ch37.
 andreaswallberg
andreaswallberg
All 25 comments
Hi there! Great to hear that you are using Scout for teaching! We have done that a few times ourselves, mostly with adapted clinical cases or spiked GiB reference samples. We might be able to share some of the latter publicly, but haven't got a good sharing system up for that yet. Tentatively looking at figshare now. It would be fun to look at your solution later! Are you running it on UPPMAX or somewhere else?
Cases are always loaded, deleted etc via the CLI. The intention being that in a production facility tasks like this is usually automated, or at least performed by informatics personell who strongly prefer the repeatability of a CLI over clicking. 😸
Yes, basically a pedigree and filenames to the vcf files you actually want loaded. Some of the meta-data files are redundant, reflecting different (versions of) pipelines and their approaches to encoding the same info. I would suggest looking att the demo cases Scout load yaml files - e g (https://github.com/Clinical-Genomics/scout/blob/master/scout/demo/cancer.load_config.yaml or https://github.com/Clinical-Genomics/scout/blob/master/scout/demo/643594.config.yaml) - and trimming away everything that you don't have or understand. It will likely work - or warn accordingly. We can try to help you from there!
In principle, GRCh37 and GRCh38 should work, but take care to specify for each case if you load 38 since 37 is default in some places (human_genome_build: 38 in the case load config yaml). Solna run mostly pure 37 instances, but e.g. Lund have mixed and mostly 38.
 dnil
on 24 Aug 2020
dnil
on 24 Aug 2020
Have you tried the admin guide in the docs dir (mirrored on e.g. http://www.clinicalgenomics.se/scout/)? I realise it may not be clear on all counts. Please keep asking for what's missing!
dnil
on 24 Aug 2020
(E.g. http://www.clinicalgenomics.se/scout/admin-guide/load-config/#minimal-config ☺️)
dnil
on 24 Aug 2020
Hi again!
I have no experience running web servers on UPPMAX, so have this set up on hardware that I control myself. I have a machine with 16 GB RAM and four (Celeron) cores and a new SSD. Do you think this machine has enough power?
With regards to loading example data, if you have something sitting around on disk from previous classes, we are interested. Out main issue and time-sink is to generate data which actually looks like it comes from related individuals. Any such data is welcome (we can put our own causative variants in), as long as there are no privacy concerns.
I will take a look at the examples! Many thanks!
andreaswallberg
on 24 Aug 2020
Depends a bit on how big your class is.. 😉 But sure, dev from small laptops work fine, and as long as you don't plan to load thousands of whole genomes I doubt you need to worry much. Having a machine of your own is in many ways ideal. We have a slightly dated AWS image that we probably should keep updated for e.g. teaching purposes; see e.g. https://github.com/Clinical-Genomics/scout/issues/1799.
I'll see what I can do about public test data, but we have talked about that internally many times, so don't expect any miracles! 😊
dnil
on 24 Aug 2020
Thanks again!
Two points:
- Lets say we have 15 students that will log into the server and muck around and do stuff at the same time. If we stick to a dataset like 643594 with three individuals and about 1000 variants, can we expect Scout to work or to tank?
- A simple way of generating new trio data is to just reuse the three individuals ADM1059A1, ADM1059A2, ADM1059A3 from from 643594, remove whatever strongly associated variants you have between phenotypes and genotypes and put in our own, right? This way, we can work from data that we know works with the platform, and which is not overly complex. However, does the data indeed represent a pedigree of related individuals, or was it just structured this way for the example?
andreaswallberg
on 24 Aug 2020
Backing up a step, when running scout setup database , is normal and expected that scouts prints that very many genes from various resources do not exist?
I wonder because downstream of this, I have problems running
scout load panel scout/demo/panel_1.txt
It also reports various missing genes.
andreaswallberg
on 24 Aug 2020
Started all over and rebuilt the database and loaded the panel and example case without issues. Maybe just some temporary external server hickup?
andreaswallberg
on 24 Aug 2020
An update, after sorting out some issues in #2040 , Scout and the database is now up again.
I loaded the test example in two steps (note: with panel-id):
scout load panel --panel-id panel1 scout/demo/panel_1.txt
scout load case scout/demo/643594.config.yaml
I can now click my way in to the following screen:
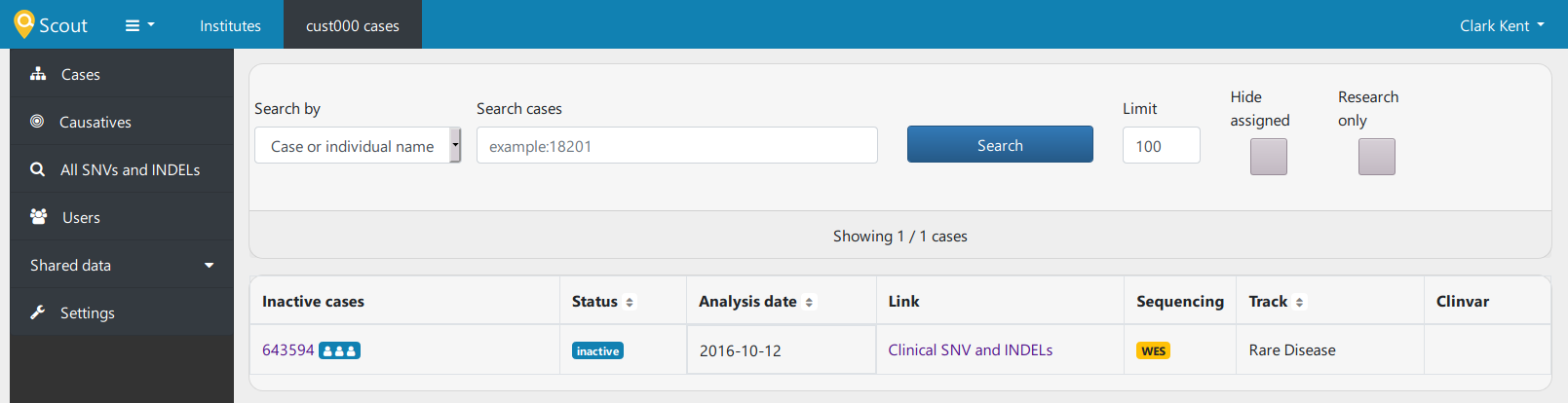
If I click "Clinical SNV and INDELs" under Link, I am presented with some interesting genes and stuff, but if I click "643594" under inactive cases, I am not taken to the nice pedigree display that I saw in the demo, but I get an "Internal Server Error", with the following messages at the end of the log:
File "/home/scoutuser/.local/lib/python3.8/site-packages/flask/helpers.py", line 370, in url_for
return appctx.app.handle_url_build_error(error, endpoint, values)
File "/home/scoutuser/.local/lib/python3.8/site-packages/flask/app.py", line 2216, in handle_url_build_error
reraise(exc_type, exc_value, tb)
File "/home/scoutuser/.local/lib/python3.8/site-packages/flask/_compat.py", line 39, in reraise
raise value
File "/home/scoutuser/.local/lib/python3.8/site-packages/flask/helpers.py", line 357, in url_for
rv = url_adapter.build(
File "/home/scoutuser/.local/lib/python3.8/site-packages/werkzeug/routing.py", line 2179, in build
raise BuildError(endpoint, values, method, self)
werkzeug.routing.BuildError: Could not build url for endpoint 'report.report' with values ['level', 'panel_name', 'sample_id']. Did you mean 'cases.hpoterms' instead?
It seems like something is up with reporting and hpoterms.
Is some other resource meant to be loaded before this works?
Am I missing some set of terms (e.g. Phenomizier, which I do not have logins for)?
andreaswallberg
on 25 Aug 2020
scout view hpo
reports that:
scout.commands.view.hpo[155551] INFO Found 15530 terms
andreaswallberg
on 25 Aug 2020
What if you run the server without gunicorn?
 northwestwitch
on 25 Aug 2020
northwestwitch
on 25 Aug 2020
Nah, you can go just fine without phenomizer, chanjo etc as long as they are not enabled in your config. I wonder if we have tested 3.8 lately - I think the automatic test matrix is 3.6 and 3.7 only?
dnil
on 25 Aug 2020
It works without gunicorn! I can load the broken link with:
scout serve -h 0.0.0.0 -p XXXXX
If the performance is expected to be acceptable with 15-20 users w/o gunicorn, I could settle with that. However, is there a way to load my key and cert w/o gunicorn, so that I can still use HTTPS (helps a lot for writing the lab instructions if this is consistent)?
andreaswallberg
on 25 Aug 2020
I wonder if we have tested 3.8 lately
I have 3.8 on my local instance and it works
northwestwitch
on 25 Aug 2020
By not enabled, should I comment them out, or keep them as they are in the example?
andreaswallberg
on 25 Aug 2020
There are also unconfigured lines for SQLALCHEMY in the my config.
andreaswallberg
on 25 Aug 2020
Commented out, or removed - if I understood what file you copied, that would still contain lines for e.g. PhenomizerUser and password, as well as Chanjo etx.
The latter SQLALCHEMY lines are for chanjo, and likely what is acting up. I would again suggest removing or commenting out anything that you are not sure you have set up already! 😅
dnil
on 25 Aug 2020
(And on our to-do we would add providing a minimal production setup server config!)
dnil
on 25 Aug 2020
That is correct, with it set to this in the config:
SQLALCHEMY_DATABASE_URI = '???'
It crashes. With it commented out, it works :-)
So a minimal config works fine without Phenomizer or SQLALCHEMY. Is there a particular function/experience in Scout that becomes degraded without Phenomizer?
andreaswallberg
on 25 Aug 2020
By not enabled, should I comment them out, or keep them as they are in the example?
I don't get it, what lines should you comment out?
scout serve -h 0.0.0.0 -p XXXXX
In general I just run scout serve
Our production server uses nginx to proxy the service. I'm copying from our setup manual:
The web service is run on http://localhost:8080. We let _NGINX_ handle the specifics of which domain the web service is available on as well as encypting traffic over SSL/HTTPS.
NGINX
To make life easy for users we proxy each sevice behind _NGINX_ so we can use pretty URLs such as scout.scilifelab.se.
- everything is behind
https://managed by _NGINX_ with SLL certificates that SciLifeLab IT provides, requests on port 80 are automatically forwarded to port 443 - IP-based access rules are setup for each web service individually in the _NGINX_ configuration
- for each subdomain we have a separate config file under
clinical-db:/etc/nginx/conf.d/{SUBDOMAIN}.conf
Each subdomain config contains the following (abbreviated) setup:
server {
server_name {SUBDOMAIN}.scilifelab.se
location / {
proxy_pass http://localhost:{PORT}; # port where the server runs
proxy_set_header Host $host;
proxy_set_header X-Real-IP $remote_addr;
proxy_set_header X-Forwarded-For $proxy_add_x_forwarded_for;
proxy_set_header X-Forwarded-Proto $scheme;
}
}
Web service run on http://localhost:XXXX. _NGINX_ will handle forwarding requests and setting the correct headers to make it all work. Since _NGINX_ also handles the https part - individual web services don't need to worry about that!
Whenever you update access rules or other settings you need to restart _NGINX_ for the changes to take effect. To restart _NGINX_, run:
sudo /sbin/service nginx restart
SQLALCHEMY
Leave it out, it's for connecting to another database (chanjo) that handles the coverage. You don't need it at this stage
northwestwitch
on 25 Aug 2020
I used the example configuration over at https://github.com/Clinical-Genomics/scout to make my config.
This example contains a few lines that make it instructions inconsistent or confusing to get going for a beginner:
MONGO_DBNAME = 'scoutTest' <-- scoutTest is never created when initiating the database. It is called 'scout' in mongodb.
To use simple login, the whole section with Google OAth need to be taken out or commented.
It is unclear to an outsider whether these '???' specifications are interpreted as invalid and discarded by scout, or set to real values internally.
# enable Phenomizer gene predictions from phenotype terms
PHENOMIZER_USERNAME = '???'
PHENOMIZER_PASSWORD = '???'
# enable Chanjo coverage integration
SQLALCHEMY_DATABASE_URI = '???'
They seem to be accepted and configure the server to use those strings. Having Phenomizer set to '???' have not triggered a crash to me (yet), but SQLALCHEMY_DATABASE_URI with '???' did.
By removing them or commenting them out, all the things that I have interacted with on the web have worked, so far.
So as @dnil said, very basic config for gunicorn with these fixed would avoid those pitfalls for a beginner like me.
andreaswallberg
on 25 Aug 2020
By removing them or commenting them out, all the things that I have interacted with on the web have worked, so far.
Ah ok, I see. If you haven't set up the system for advances settings then just omit them
northwestwitch
on 25 Aug 2020
Yep. The issue, I have no idea what settings are advanced or not when setting this up for the first time :-D
andreaswallberg
on 25 Aug 2020
Yep. The issue, I have no idea what settings are advanced or not when setting this up for the first time :-D
You haven't set up google OAuth so omit, same for phenomizer, same for SQLALCHEMY_DATABASE_URI. Just omit the lot of them
northwestwitch
on 25 Aug 2020
For reference, I added a few lines on this in the docs (#2046). Since it seems you are up and running for the moment, so I'll close the thread, but feel free to reopen/open new ones! Its really useful to have other eyes look at this.
dnil
on 25 Aug 2020
Related issues
 ielvers
·
3Comments
ielvers
·
3Comments
 heronikdin
·
4Comments
heronikdin
·
4Comments
 hassanfa
·
3Comments
hassanfa
·
3Comments
 1ctw
·
5Comments
dnil
·
3Comments
1ctw
·
5Comments
dnil
·
3Comments