Picongpu: first run on the cluster
I am trying to get my PIConGPU model running on a cluster for the very first time. So far I managed to transfer my files there and compile, everything using Spack. All fine.
Now I have very limited knowldge about clusters and Slurm. I understand that instead of the typical .cfg file I need to launch a .tpl file and this will schedule the job but I have two questions:
1) do I need to know something about the architecture of the cluster so I can modify the template for the .tpl files that you rpovide. For example https://github.com/ComputationalRadiationPhysics/picongpu/blob/dev/etc/picongpu/hemera-hzdr/gpu.tpl ?
Here is the .tpl file which I could use.
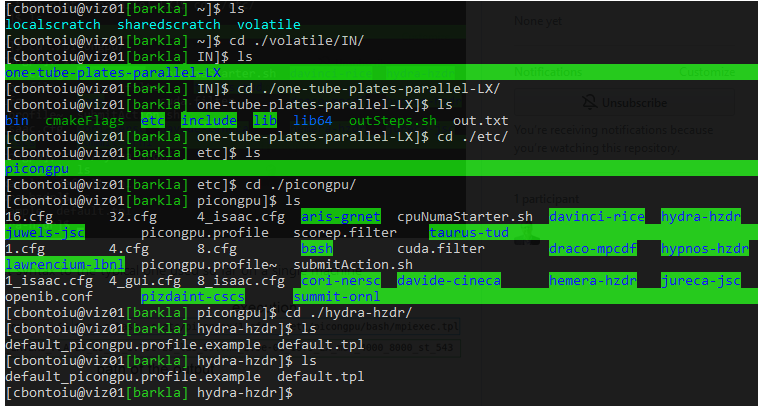
2) what would be equivalent syntax to the typical one used so far on a single machine
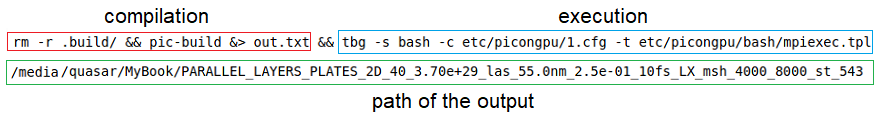
Is the .cfg file still needed (I assume so) and I only need to target a different file than mpiexec.tpl or what else should I do?
I would appreciate a bit of guidance as this is the very firrst contact with HPC for me. Thank you very much.
 cbontoiu
cbontoiu
All 129 comments
Hello @cbontoiu .
.cfg files have run-time parameters of PIConGPU (grid size, plugins, etc.). These do not depend on whether you run on a cluster with multiple nodes, or just on a local computer. The only difference is values of the TBG_devices_{x|y|z} variables: these mean how many MPI processes you want to use for decomposition along x, y, z. The total number of processes is the multiple of those three. As you probably noted, our examples normally come with a set of .cfg files for different number of processes and appropriate grid sizes.
 sbastrakov
on 9 Jun 2020
sbastrakov
on 9 Jun 2020
Regarding .tpl file, I think you would indeed benefit from making one, to be reused for all your runs on that cluster. This file describes the job submission part of a cluster: batch system (slurm, etc.), partition name, number of CPU cores and amount of memory per node. I think you could indeed take the linked file for Hemera and modify it using the cluster details, as it is also for SLURM and the variables hopefully have rather telling names. Please do not hesitate to ask questions.
sbastrakov
on 9 Jun 2020
To add to my previous message, after you produced some .tpl file, run PIConGPU on the cluster as shown here. You could keep tbg -s sbatch, then for the -c argument provide the path to your .cfg file, and for the -t argument - the path to your .tpl file. The last parameter.is the output directory. This is specific to your cluster, there should be a directory on large storage, often but not always denoted by environment variable $SCRATCH, please contact documentation of the cluster for it.
sbastrakov
on 9 Jun 2020
@cbontoiu Great to hear that you switched to a cluster. As @sbastrakov already mentioned, feel free to ask questions at any time. If you could give us the name of the cluster, we might even help a bit in determining the relevant parameters to change if they are available online (most research clusters publish their configuration online).
 PrometheusPi
on 9 Jun 2020
PrometheusPi
on 9 Jun 2020
The cluster is Barkla at Liverpool University.
https://www.liverpool.ac.uk/csd/advanced-research-computing/facilities/high-performance-computing/
I am tring to follow your suggestions. I currently modify the gpu.tpl file from hemera-hzdr. I see a gpu_picongpu.profile.example file there and it is supposed to load some modules. I assume this is done for me using Spack by source volatile/src/spack/share/spack/setup-env.sh which is automatically called once i connect to the cluster via the .bashrc execution.
cbontoiu
on 9 Jun 2020
Yes, so the environment that we configure in gpu_picongpu.profile.example should be taken care of with spack in your case. And if you made it compile, probably this part works fine. So, at least in the ideal case, you need to only make a .tpl file for this system.
sbastrakov
on 9 Jun 2020
Well, I tried first k20.tpl without modifuing anything there and obtained
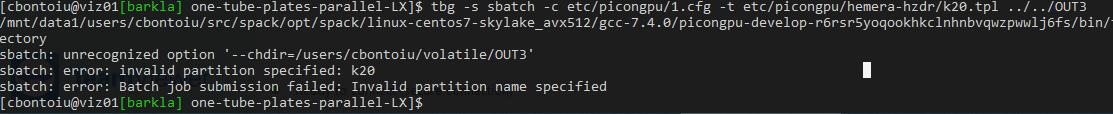
I think it needs some adjusments for my cluster

I also modified my 1.cfg file adding more gpus for my 2D simulation.
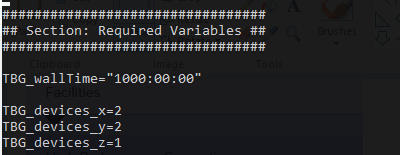 ;
;
Sould there be an exact match between the numbers of GPUs used in the .cfg and .tpl files? Should they be what physically exist or Slurm adjusts this?
cbontoiu
on 9 Jun 2020
And using the gpu.tpl without any modifications gave me an error about a folder which cannot be created I guess and a problem with the wall time

cbontoiu
on 9 Jun 2020
Hello @cbontoiu .
Regarding the first question, the issue is that Barkla's partition names are (naturally) different from Hemera's. So the partition name has to be changed according to what the cluster has. It can be set in this line. The names of partitions should be part of user documentation for the cluster. There are some names here, but I guess there is some other page explaining what partitions there are and what hardware is there. Particularly, GPU nodes are often in their own partition, so you would need to find out in the documentation or via cluster support.
Then you would need to adjust at least the number of GPUs per node, amount of memory (RAM, not storage) per GPU and per node, and number of CPUs cores per each GPU. All of these should be part of the documentation. I do not see it on the linked overview page, but there must be more detailed documentation file or website, maybe it is only for users.
Regarding the .tpl vs. .cfg files. The former describes the machine itself, so e.g. the number of GPUs per node or amount of memory is how much does the machine have. The latter concerns your particular simulation setup. So if e.g. there were 2 GPUs per node on a cluster and you request 4 devices like in the given example, it's fine, tbg will ask for 2 nodes to run this simulation.
sbastrakov
on 10 Jun 2020
For the "Requested time limit is invalid". There are normally limitations on all clusters for max time per one job, so 1000 hours is likely far beyond those. Please consult the documentation or support to find out the limit. Otherwise your jobs won't start as the wall time parameter is illegal. As a side note, PIConGPU should be much faster on GPUs (depends on hardware, but could expect an order of magnitude faster than CPUs of the same era), so perhaps you won't need that much time.
sbastrakov
on 10 Jun 2020
Concerning the chdir error, I am not sure whether it's because the path was wrong or the option is somehow not supported on the machine (in this case we can work around it rather easily). I think, as a patch you could try just removing this line from the .tpl.
sbastrakov
on 10 Jun 2020
Regarding the chdir issue: The SLURM is probably not at a current version, thus just replace --chdir by --workdir should solve the issue. (We had to update to the new interface for a couple of clusters already, see #3181 and #3123)
PrometheusPi
on 11 Jun 2020
I got an example of slurm file from the admins of the Barkla cluster as the one below. I am still trying to merge the picongpu one with this one below. I use Spack so all modules are loaded before using this file.
#!/bin/bash -l
# Use the current working directory
#SBATCH -D ./
# Use the current environment for this job
#SBATCH --export=ALL
# Define job name
#SBATCH -J TFGPU
# Define a standard output/error file. When the job is running, %N will be replaced by the name of
# the first node where the job runs, %j will be replaced by job id number.
#SBATCH -o tensorflow.%N.%j.out
# Request the GPU partition
#SBATCH -p gpu
# Request the number of GPUs to be used (if more than 1 GPU is required, change 1 into Ngpu, where Ngpu=2,3,4)
#SBATCH --gres=gpu:1
# Request the number of nodes
#SBATCH -N 1
# Request the number of CPU cores. (There are 24 CPU cores and 4 GPUs on the GPU node,
# so please request 6*Ngpu CPU cores, i.e., 6 CPU cores for 1 GPU, 12 CPU cores for 2 GPUs, and so on.
# User may request more CPU cores for jobs that need very large CPU memory occasionally,
# but the number of CPU cores should not be greater than 6*Ngpu+5. Please email hpc-support if you need to do so.)
#SBATCH -n 6
# Set time limit in format a-bb:cc:dd, where a is days, b is hours, c is minutes, and d is seconds.
#SBATCH -t 1-00:00:00
# Request the memory on the node or request memory per core
# PLEASE don't set the memory option as we should use the default memory which is based on the number of cores
##SBATCH --mem=90GB or #SBATCH --mem-per-cpu=9000M
# Insert your own username to get e-mail notifications (note: keep just one "#" before SBATCH)
##SBATCH --mail-user=<username>@liverpool.ac.uk
# Notify user by email when certain event types occur
#SBATCH --mail-type=ALL
#
# Set your maximum stack size to unlimited
ulimit -s unlimited
# Set OpenMP thread number
export OMP_NUM_THREADS=$SLURM_NTASKS
# Load tensorflow and relevant modules
#module load libs/scipy
#module load libs/pandas
#module load apps/tensorflow/1.6.0
module load apps/anaconda3/5.2.0
#use source activate gpu to get the gpu virtual environment
source activate gpu
# List all modules
module list
echo =========================================================
echo SLURM job: submitted date = `date`
date_start=`date +%s`
hostname
echo "CUDA_VISIBLE_DEVICES : $CUDA_VISIBLE_DEVICES"
echo "GPU_DEVICE_ORDINAL : $GPU_DEVICE_ORDINAL"
echo "Running GPU jobs:"
echo "Running GPU job tf_test_graph.py:"
python tf_test_graph.py
echo "Running GPU job tf_benchmark_sky_alexnet.py:"
python tf_benchmark_sky_alexnet.py
#deactivate the gpu virtual environment
source deactivate gpu
date_end=`date +%s`
seconds=$((date_end-date_start))
minutes=$((seconds/60))
seconds=$((seconds-60*minutes))
hours=$((minutes/60))
minutes=$((minutes-60*hours))
echo =========================================================
echo SLURM job: finished date = `date`
echo Total run time : $hours Hours $minutes Minutes $seconds Seconds
echo =========================================================
That is great. Only slight adjustments to hzdr-hermera/gpu.tpl are needed. The system looks very similar: 6 cores per, 4 GPUs per node, partition name "gpu". They only request to not set memory manually.
....
-# PIConGPU batch script for hemera's SLURM batch system
+# PIConGPU batch script for Barkla's SLURM batch system
#SBATCH --partition=!TBG_queue
#SBATCH --time=!TBG_wallTime
# Sets batch job's name
#SBATCH --job-name=!TBG_jobName
#SBATCH --nodes=!TBG_nodes
#SBATCH --ntasks=!TBG_tasks
#SBATCH --ntasks-per-node=!TBG_gpusPerNode
#SBATCH --mincpus=!TBG_mpiTasksPerNode
#SBATCH --cpus-per-task=!TBG_coresPerGPU
-#SBATCH --mem=!TBG_memPerNode
#SBATCH --gres=gpu:!TBG_gpusPerNode
#SBATCH --mail-type=!TBG_mailSettings
#SBATCH --mail-user=!TBG_mailAddress
-#SBATCH --chdir=!TBG_dstPath
+#SBATCH -D=!TBG_dstPath
#SBATCH -o stdout
#SBATCH -e stderr
## calculations will be performed by tbg ##
.TBG_queue="gpu"
# settings that can be controlled by environment variables before submit
.TBG_mailSettings=${MY_MAILNOTIFY:-"NONE"}
.TBG_mailAddress=${MY_MAIL:-"[email protected]"}
.TBG_author=${MY_NAME:+--author \"${MY_NAME}\"}
.TBG_profile=${PIC_PROFILE:-"~/picongpu.profile"}
# number of available/hosted GPUs per node in the system
.TBG_numHostedGPUPerNode=4
# required GPUs per node for the current job
.TBG_gpusPerNode=`if [ $TBG_tasks -gt $TBG_numHostedGPUPerNode ] ; then echo $TBG_numHostedGPUPerNode; else echo $TBG_tasks; fi`
-# host memory per gpu
-.TBG_memPerGPU="$((378000 / $TBG_gpusPerNode))"
-# host memory per node
-.TBG_memPerNode="$((TBG_memPerGPU * TBG_gpusPerNode))"
# number of cores to block per GPU - we got 6 cpus per gpu
# and we will be accounted 6 CPUs per GPU anyway
.TBG_coresPerGPU=6
# We only start 1 MPI task per GPU
.TBG_mpiTasksPerNode="$(( TBG_gpusPerNode * 1 ))"
# use ceil to caculate nodes
.TBG_nodes="$((( TBG_tasks + TBG_gpusPerNode - 1 ) / TBG_gpusPerNode))"
## end calculations ##
echo 'Running program...'
...
should do the trick.
PrometheusPi
on 12 Jun 2020
Thanks for such a quick reply! I am trying now. So # is not a comment for slurm?
cbontoiu
on 12 Jun 2020
@cbontoiu #SLURM is a slurm command - because the file is in principle a bash script, SLURM command are hidden behind bash comments. Thus a line in the script like #SBATCH -p gpu is equal to sbatch -p gpu <the rest of the script>
PrometheusPi
on 12 Jun 2020
@PrometheusPi
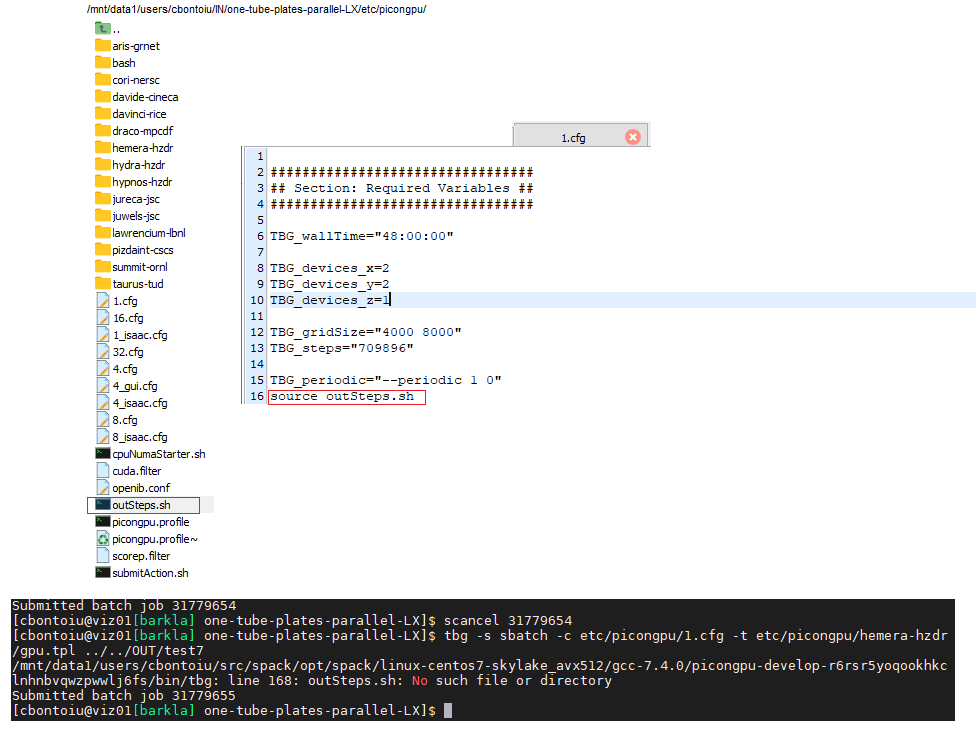
It seems I can schedule the job now! Great and thanks! I also learnt how to stop the job, but still my outSteps.sh file cannot be found/read even after moving it in the same folder as the 1.cfg file from which it is called, and this is a new behaviour only with the current cluster-specific commands.
cbontoiu
on 12 Jun 2020
The bash script that is executed and can not find the outSteps.sh file is executed from within the directory you defined via #SBATCH -D .... You can either place outSteps.sh in this directory or you give an absolute path to the script.
PrometheusPi
on 13 Jun 2020
Thank you all! It was easier than I thought. I am grateful.
cbontoiu
on 14 Jun 2020
@cbontoiu I am glad it worked that easily :+1: - Are you willing to submit a pull request that adds a *.tpl and *_picongpu.profile.example for the Barkla cluster via a pull request?
PrometheusPi
on 15 Jun 2020
@cbontoiu I am glad it worked that easily 👍 - Are you willing to submit a pull request that adds a
*.tpland*_picongpu.profile.examplefor the Barkla cluster via a pull request?
Yes, I will try to do that. I am only waiting for the first results to appear, because my job was pending for two days and I am not sure what happens there on the cluser. I asked some help from the admins and hoepfully they will review my tpl file as well.
It seems that I am limited to 4 GPUs working on a single node. I'm not am sure if it is ever possible to use 2 or more nodes. By that way my tpl file containes a few TBG variables (lines 3-15). Among them I cannot understand where $TBG_tasks is defined. Also, what can I do with lines 25-28 to make sure I get notified when my job starts?
#!/bin/bash -l
# PIConGPU batch script for Barkla's SLURM batch system
#SBATCH --partition=!TBG_queue
#SBATCH --time=!TBG_wallTime
# Sets batch job's name
#SBATCH --job-name=!TBG_jobName
#SBATCH --nodes=!TBG_nodes
#SBATCH --ntasks=!TBG_tasks
#SBATCH --ntasks-per-node=!TBG_gpusPerNode
#SBATCH --mincpus=!TBG_mpiTasksPerNode
#SBATCH --cpus-per-task=!TBG_coresPerGPU
#SBATCH --gres=gpu:!TBG_gpusPerNode
#SBATCH --mail-type=!TBG_mailSettings
#SBATCH --mail-user=!TBG_mailAddress
#SBATCH -D=!TBG_dstPath
#SBATCH -o stdout
#SBATCH -e stderr
## calculations will be performed by tbg ##
.TBG_queue="gpu"
# settings that can be controlled by environment variables before submit
.TBG_mailSettings=${MY_MAILNOTIFY:-"NONE"}
.TBG_mailAddress=${MY_MAIL:-"[email protected]"}
.TBG_author=${MY_NAME:+--author \"${CBONTOIU}\"}
.TBG_profile=${PIC_PROFILE:-"~/picongpu.profile"}
# number of available/hosted GPUs per node in the system
.TBG_numHostedGPUPerNode=4
# required GPUs per node for the current job
.TBG_gpusPerNode=`if [ $TBG_tasks -gt $TBG_numHostedGPUPerNode ] ; then echo $TBG_numHostedGPUPerNode; else echo $TBG_tasks; fi`
# number of cores to block per GPU - we got 6 cpus per gpu
# and we will be accounted 6 CPUs per GPU anyway
.TBG_coresPerGPU=6
# We only start 1 MPI task per GPU
.TBG_mpiTasksPerNode="$(( TBG_gpusPerNode * 1 ))"
# use ceil to caculate nodes
.TBG_nodes="$((( TBG_tasks + TBG_gpusPerNode - 1 ) / TBG_gpusPerNode))"
## end calculations ##
echo 'Running program...'
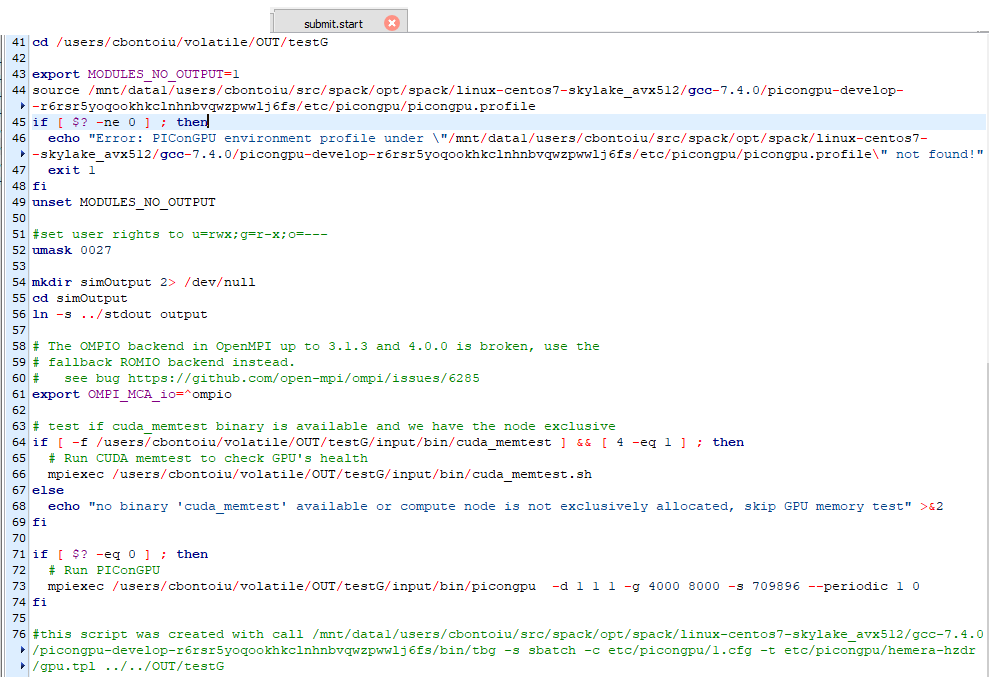
cd !TBG_dstPath
export MODULES_NO_OUTPUT=1
source !TBG_profile
if [ $? -ne 0 ] ; then
echo "Error: PIConGPU environment profile under \"!TBG_profile\" not found!"
exit 1
fi
unset MODULES_NO_OUTPUT
#set user rights to u=rwx;g=r-x;o=---
umask 0027
mkdir simOutput 2> /dev/null
cd simOutput
ln -s ../stdout output
# The OMPIO backend in OpenMPI up to 3.1.3 and 4.0.0 is broken, use the
# fallback ROMIO backend instead.
# see bug https://github.com/open-mpi/ompi/issues/6285
export OMPI_MCA_io=^ompio
# test if cuda_memtest binary is available and we have the node exclusive
if [ -f !TBG_dstPath/input/bin/cuda_memtest ] && [ !TBG_numHostedGPUPerNode -eq !TBG_gpusPerNode ] ; then
# Run CUDA memtest to check GPU's health
mpiexec !TBG_dstPath/input/bin/cuda_memtest.sh
else
echo "no binary 'cuda_memtest' available or compute node is not exclusively allocated, skip GPU memory test" >&2
fi
if [ $? -eq 0 ] ; then
# Run PIConGPU
mpiexec !TBG_dstPath/input/bin/picongpu !TBG_author !TBG_programParams
fi
Hello @cbontoiu .
The line TBG_numHostedGPUPerNode=4 means that the cluster has 4 GPUs per node on the corresponding partition. How many nodes you use depends on the number of MPI ranks (equal to number of GPUs for this case) requested from your .cfg file with TBG_devices_x/y/z variables. This number is set to TBG_tasks variable at the end of each .cfg file (you are not supposed to modify its equation). So then the number of nodes is calculated as the upper integer part of the ratio as you can see in the TBG_nodes expression in the .tpl file
Regarding notifications, you can manage it by setting up the MY_MAILNOTIFY variable between these two lines:
# settings that can be controlled by environment variables before submit
.TBG_mailSettings=${MY_MAILNOTIFY:-"NONE"}
E.g.
# settings that can be controlled by environment variables before submit
export MY_MAILNOTIFY="ALL"
.TBG_mailSettings=${MY_MAILNOTIFY:-"NONE"}
to get all notifications. Please see the other options here.
sbastrakov
on 15 Jun 2020
Also, generally for testing that the code runs on a new machine you may want to first submit some small jobs for e.g. 1 GPU and 10 minutes using your reduced setup, or a standard example. The queue waiting time should normally be much quicker for this case. And only after everything is setup properly run your real setups.
sbastrakov
on 15 Jun 2020
OK, so if I set TBG_wallTime="4:00:00" means that 1) my job will wait for 4 hours in queue and will be killed if it didn't have a chance to start or 2) my job will be killed after 4 hours from the moment it started to run?
The admin gave me this answer "Again for testing purposes, it might be sensible to set the time to a couple of hours or the minimum time required to determine if things are running correctly. The scheduler in-fills, so jobs with short run times tend to be scheduled sooner."
cbontoiu
on 15 Jun 2020
It's option 2. TBG_wallTime="4:00:00" means you request 4 hours for execution of your job. If it's not finished after 4 hours since it started, the job system will kill it. Job schedulers normally assign task priorities based on multiple factors, however wall time and number of nodes requested are normally important one.
Yes, so I would agree with the admin there. You could e.g. run the standard LWFA for 10 minutes using the existing 1.cfg in that example, just change the wall time there.
sbastrakov
on 15 Jun 2020
@cbontoiu Option 2 is correct. The job can wait indefinitely in the queue, but its run time (actual simulation time) is limited to 4 hours.
In principle SLURM tries to fill to occupy all nodes of the cluster as much as possible. Thus jobs, that run only for a short time can ideally be used by the scheduler to occupy nodes that are already reserved for another job, but were not enough nodes are yet freed. These jobs are commonly called back-fill jobs. They can run despite having low priority, because they increase the occupancy of the cluster.
Furthermore, having a short walltime on the first test is a good idea, because sometimes job scripts contain errors that lead to PIConGPU (or any other software) to not start at all but hang, thus blocking the nodes used for the duration of the walltime specified in the job script.
EDIT: Sorry for double posting - This is in principle the same answer @sbastrakov gave a few seconds ago.
PrometheusPi
on 15 Jun 2020
@PrometheusPi @sbastrakov
Hello again,
I eventually got my job running and discovered a few more problems for which I would kindly ask again you help. The situation is like this:
(1) spack is installed in my volatile/src/ folder with picongpu and all dependencies
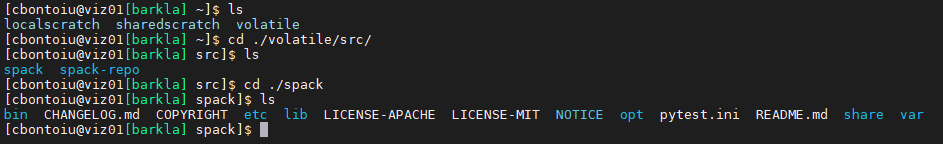
(2) my .bashrc file includes a source call to setup-env.sh;
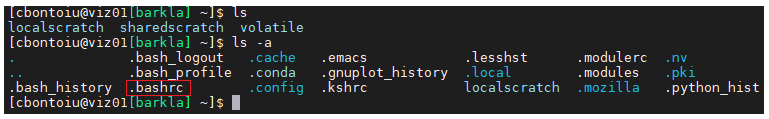
I update the path to setup-env.sh to the full path because slurm complained.
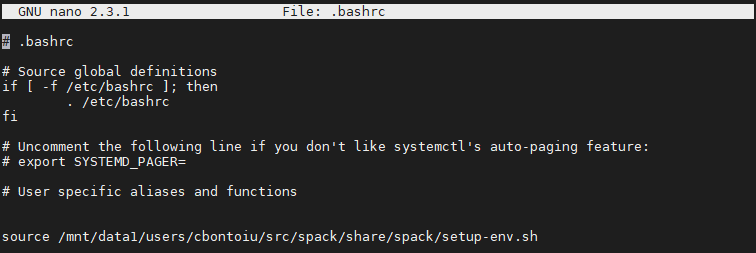
I then checked that compilation still works with rm -r .build/ && pic-build &> out.txt. All seems fine as you can check in the file below
(3) while being in the model folder I sucessfully sumbit the job with tbg -s sbatch -c etc/picongpu/1.cfg -t etc/picongpu/hemera-hzdr/gpu.tpl ../../OUT/testA
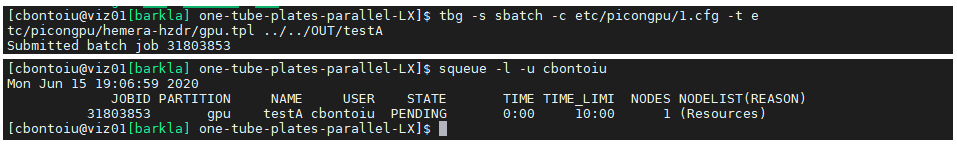
(4) there is obviously an error somewhere because the output is made of two text files
with the stderr file
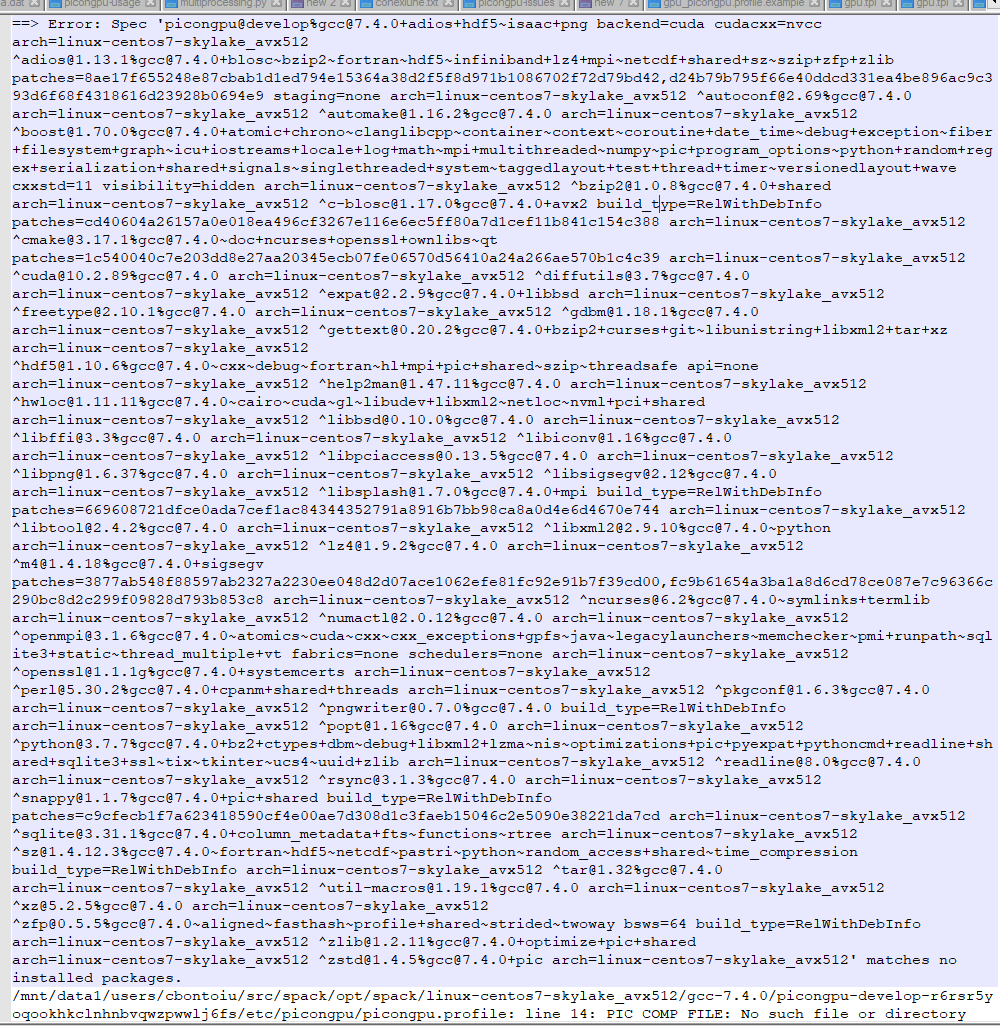
and the stdout file
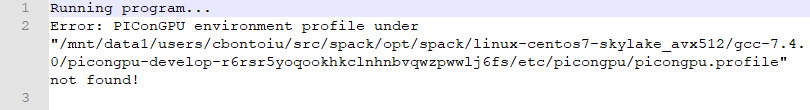
They complain about a profile but this was never really needed on a single machine with spack. It is true that the warning existed before but the model could be executed anyway. It is not the case now on the cluster.
Could you help me please? Thank you
cbontoiu
on 15 Jun 2020
I suspect the issue to arise because the .bashrc is loaded only during interactive sessions, not during other types of shell sessions, as e.g. SLURM job execution. Thus the job encounters a different environment and thus does not find the correct path. Sourcing your setup-env.sh (or your .bashrc) could solve the issue. I am a bit surprised, with module systems this is never an issue, but perhaps SPACK behaves differently. @sbastrakov could you comment on that?
PrometheusPi
on 15 Jun 2020
To me @PrometheusPi 's suggestion seems reasonable. I guess the /mnt/data1/.../setup-env.sh is not available from the compute nodes? You could figure this out by reading the docs on how the file system is organized on the cluster, or just get a node for interactive session and try out.
If so, you could move it to a shared directory accessible from both head nodes and compute ones.
sbastrakov
on 15 Jun 2020
I added a source call in the gpu.tpl file as
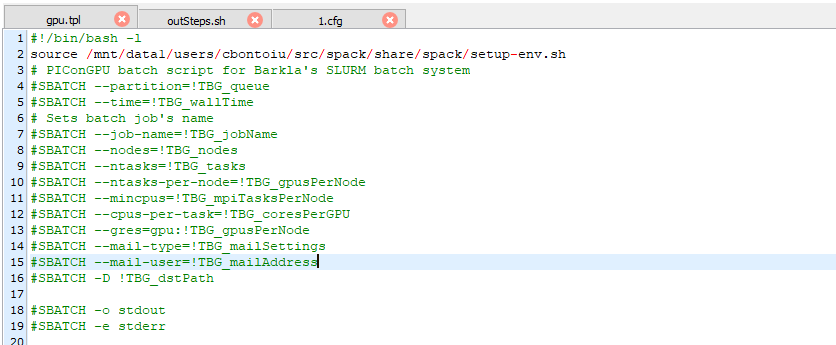
I am sure that the source command was executed, because it complained first when I gave it a wrong path. So I submitted again and I got slurm-....out as output
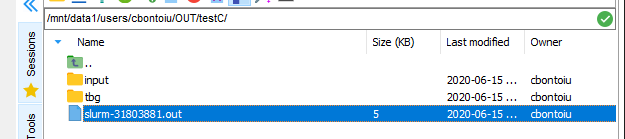
and this is what the file contains

cbontoiu
on 15 Jun 2020
So I am confused about the first error, and have no experience with spack to rely upon. Regarding the PIC_COMP_FILEerror, it seems to be in our spack file. I will provide a fix for it, for now please replace PIC_COMP_FILE with $PIC_COMP_FILE at line 14 of the picongpu.profile. However, I don't think this helps with the other error you got.
sbastrakov
on 15 Jun 2020
@sbastrakov Thank you for this suggestion. I modified the picongpu.profile as shown below but got the same error as output. I will contact the admin to see what they suggest.
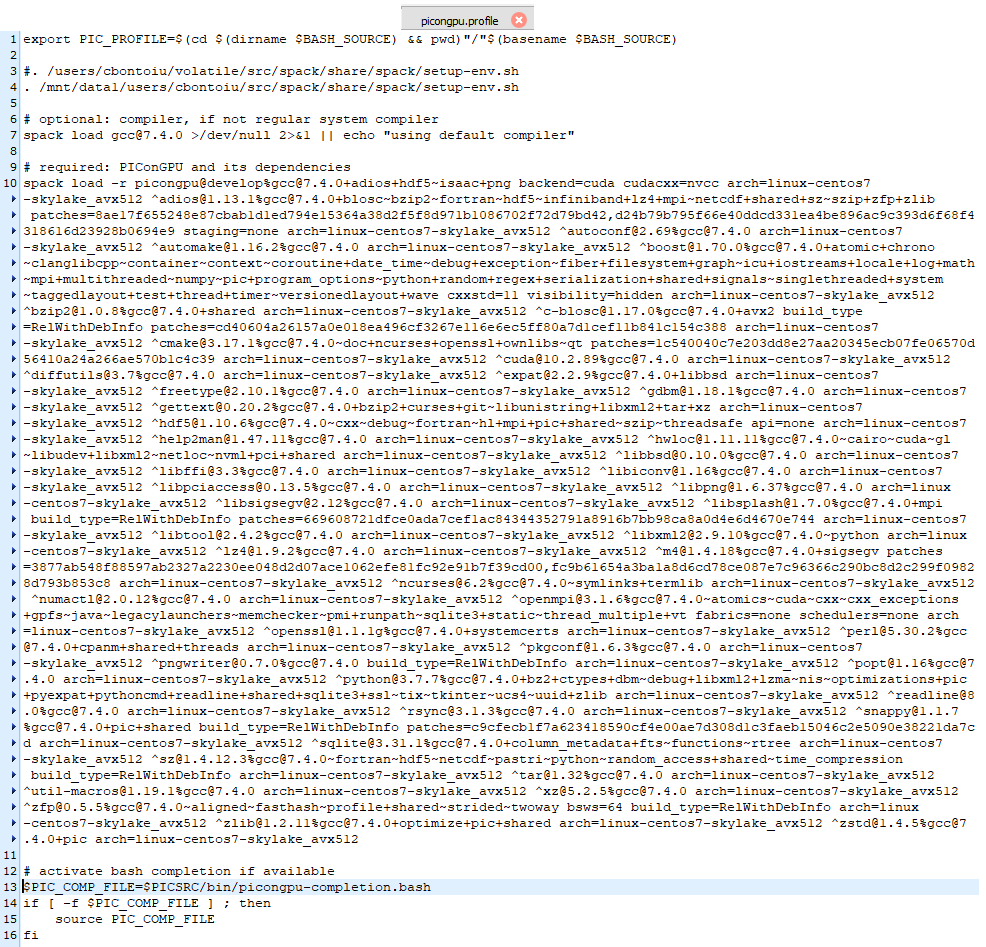
cbontoiu
on 16 Jun 2020
Hello @sbastrakov @PrometheusPi
I have an answer from the admins of the Barkla cluster. I asked them to look at this issue.
"The volatile, users and sharedscratch volumes are shared across all of the nodes. I suspect you are looking for your solution in the wrong place. The problem is not that the picongpu.profile cannot be found, but you have a typo in the file so there is an undefined file reference. This is clear from the error message you record and can be seen in the fragment below:
$PIC_COMP_FILE=$PICSRC/bin/picongpu-completion.bash
if [ -f $PIC_COMP_FILE ] ; then
source PIC_COMP_FILE
fi
That source has to be for $PIC_COMP_FILE, PIC_COMP_FILE by itself is undefined hence the error message. You are attempting an extremely complicated build. You need to look extremely carefully at all of the error messages to see what is going wrong. The downside of spack is that intermediate steps are not really human readable, which makes debugging hard. By the way, if this code is scalable, you may be better using 40 CPU cores per node with several nodes possible in a job over a single node with 4 GPUs. That only makes sense if the GPU code is more than ten times faster than the CPU code."
cbontoiu
on 16 Jun 2020
@cbontoiu so the admin found the same bug with missing $ that I pointed out in the previous message. However, you seem to have modified the variable in the wrong place: should have been inside the if statement, not at declaration.
Should be:
PIC_COMP_FILE=$PICSRC/bin/picongpu-completion.bash
if [ -f $PIC_COMP_FILE ] ; then
source $PIC_COMP_FILE
fi
Btw the fix is now merged to our spack.
Regarding the second point. I would generally agree with the admin. However, one could reasonably expect GPUs to be about 10x faster.
sbastrakov
on 16 Jun 2020
@sbastrakov
I corrected the profile and got one step further. for the first time the simOutput folder was generated but the slurm...out file indicates now a different error as you can see in the image below
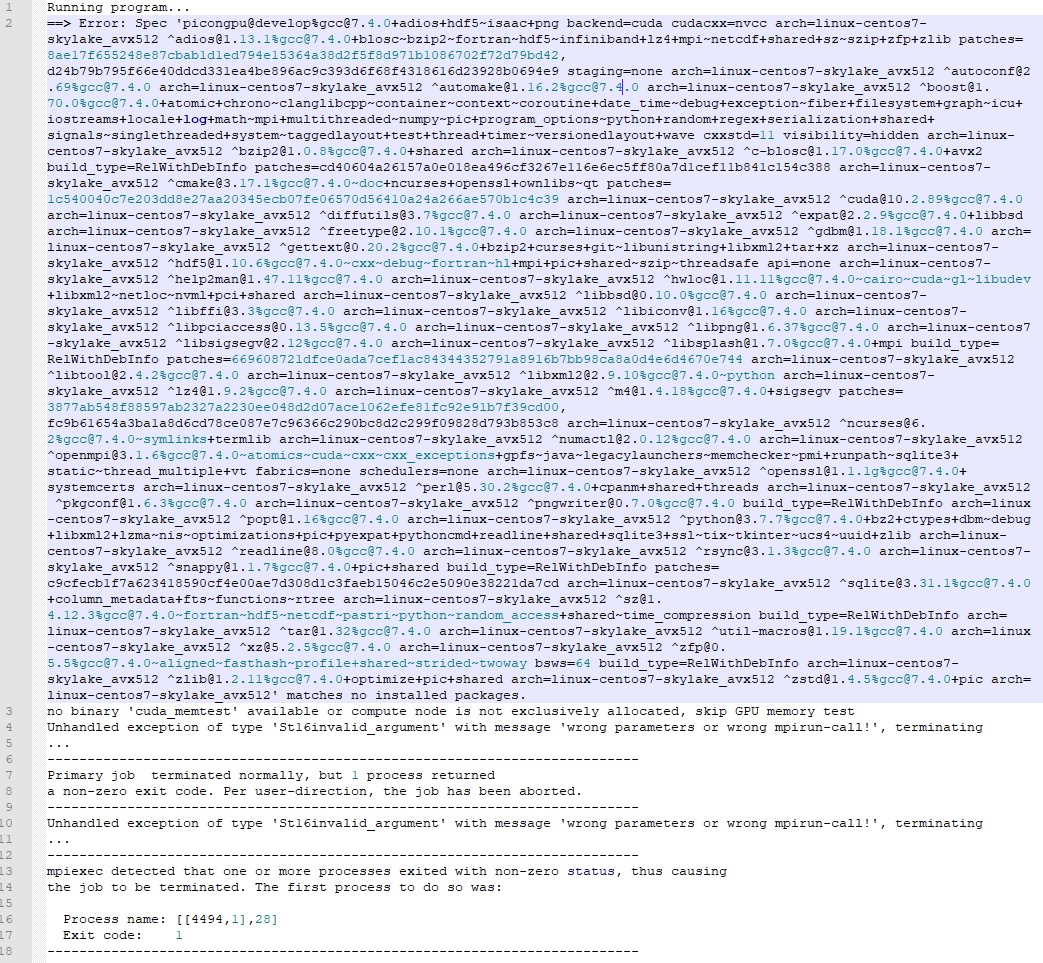
cbontoiu
on 16 Jun 2020
Okay, so that "Unhandled exception .. wrong parameters or wrong mpirun-call" definitely comes from PIConGPU itself, so it is starting and we can figure out from there. Please attach the whole tbg subdirectory from your output directory, it should be on the same level as simOutput
sbastrakov
on 16 Jun 2020
Here you have it. Thank you very much for looking into this.
cbontoiu
on 16 Jun 2020
Thanks for providing the archive. Unfortunately, I do not see what is wrong, everything looks reasonable to me. Could you check the parameters of that slurm job?
To retrieve your job history, you could use
sacct -u your_account_name_on_that_cluster -S 2020-06-16.
Then the job id from the first column of the output can be used to retrieve the information via
sacct -j that_job_id --format=User,JobID,Jobname,partition,state,time,start,end,elapsed,MaxRss,MaxVMSize,nnodes,ncpus,nodelist
(--format is without space, just how markdown shows it).
sbastrakov
on 16 Jun 2020
Hello @sbastrakov
This is the result
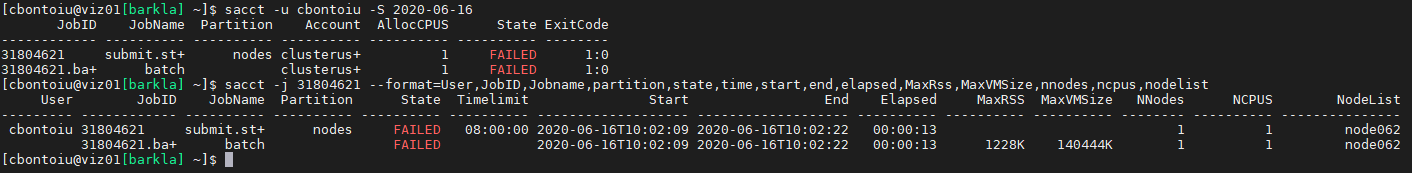
cbontoiu
on 16 Jun 2020
@cbontoiu Thanks for providing all the tbg/* files. I am a bit confused.
1.) According to tbs/submit.start you are using #SBATCH --partition=gpu the so-called gpu partition. But the jub you showed used the nodes partition. Is this the same partition and only an alias? (This is most likly not the issue, but is is a bit strange.)
2.) Why is there no cuda_memtest in /users/cbontoiu/volatile/OUT/testG/input/bin/cuda_memtest?
PrometheusPi
on 16 Jun 2020
I think poit 1) contains a hint because while everybody uses node, there is one job using gpu and this should have been my case.
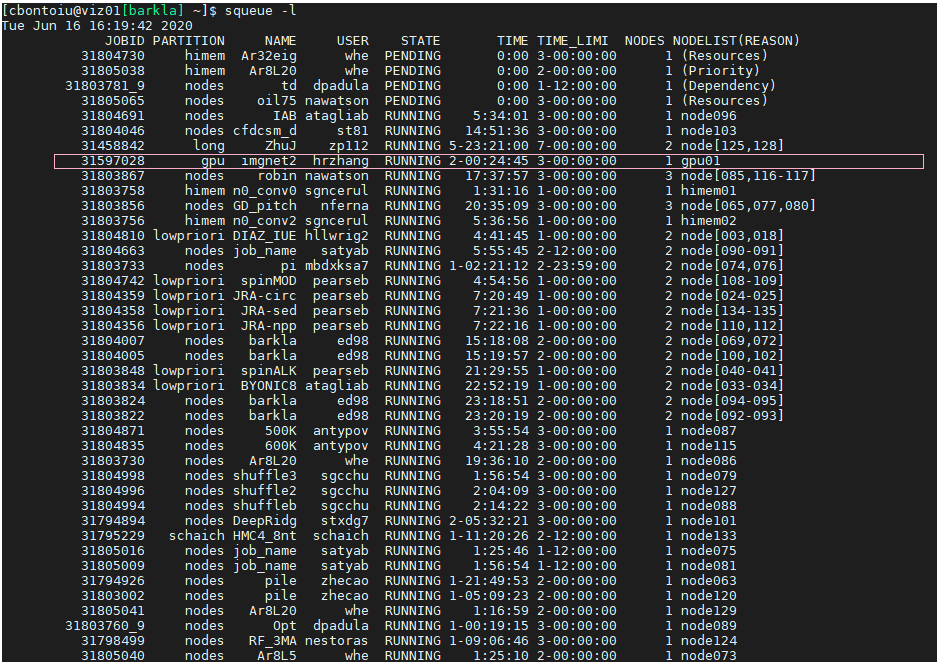
For point 2) I see the cuda_memetest.
cbontoiu
on 16 Jun 2020
One possible issue might be that you are not supposed to use mpiexec but should use something like srun (similar to taurus-zih). You might need to ask an admin for that.
(srun is practically an mpiexec or mpirun with all SLURM options included).
PrometheusPi
on 16 Jun 2020
One possible issue might be that you are not supposed to use
mpiexecbut should use something likesrun(similar to taurus-zih). You might need to ask an admin for that.
(srunis practically anmpiexecormpirunwith all SLURM options included).
OK, I will do that. So it is something related to openMPI or a version of it.
cbontoiu
on 16 Jun 2020
@sbastrakov and @PrometheusPi
I have an answer from the admin as this
"Nearly everyone uses mpirun. I would have thought your self-contained mpiexec should work. Certainly srun is not used in our system for this purpose, but I am aware that is how some systems get tight interconnection between their MPI and Slurm.
Again, mpiexec appears to have started and an error was passed to it.
Note the error message about the cuda_memtest – this was not run because you were sharing the node with other users. I don’t think this was fatal.
The nodes partition is the default partition if you leave off the –p specification. You need –p gpu to access the gpu01 node.
The error might be in the actual code invocation."
Now, do you think I need to adjust my gpu.tpl as
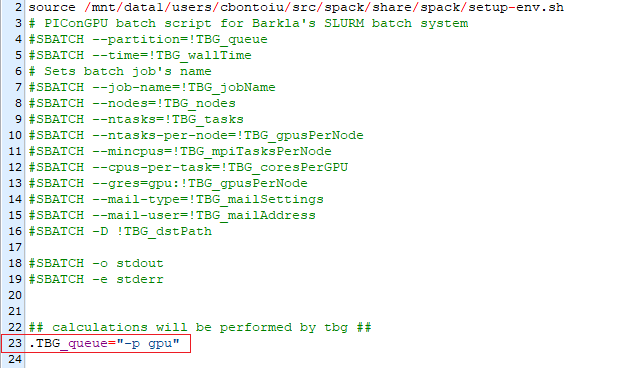
cbontoiu
on 16 Jun 2020
In a standard SLUM configuration -p and --partition are equivalent. And there you set gpu as partition name, thus this should be fine.
You should not define .TBG_queue="-p gpu" because this will lead to a SLURM command as #SBATCH --partition="-p gpu" which should not work.
Sharing a GPU node with other users might definitely be an issue with PIConGPU. You could prevent this by allocating the node #SLURM --exclusive. (@sbastrakov Or is PIConGPU now only using the assigned GPUs?)
PrometheusPi
on 16 Jun 2020
@PrometheusPi I submitted again the job
With #SLURM --exclusive added inside gpu.tpl I got a similar message as before
centos7-skylake_avx512 ^[email protected]%[email protected]+pic arch=linux-centos7-skylake_avx512' matches no installed packages.
no binary 'cuda_memtest' available or compute node is not exclusively allocated, skip GPU memory test
Unhandled exception of type 'St16invalid_argument' with message 'wrong parameters or wrong mpirun-call!', terminating
Unhandled exception of type 'St16invalid_argument' with message 'wrong parameters or wrong mpirun-call!', terminating
@cbontoiu I can not spot an error in your PIConGPU call, but to be sure, could you please adjust your submit.start to only include
mpiexec /users/cbontoiu/volatile/OUT/testG/input/bin/picongpu -d 1 1 1 -g 4000 8000 -s 709896 --periodic 1 0
and no adios output?
Then just resubmit via sbatch tbg/submit.start. Does the same error occur?
PrometheusPi
on 16 Jun 2020
@PrometheusPi
I will do that, but where can I find this file? As you see in the screenshot there is a submitAction.sh file.
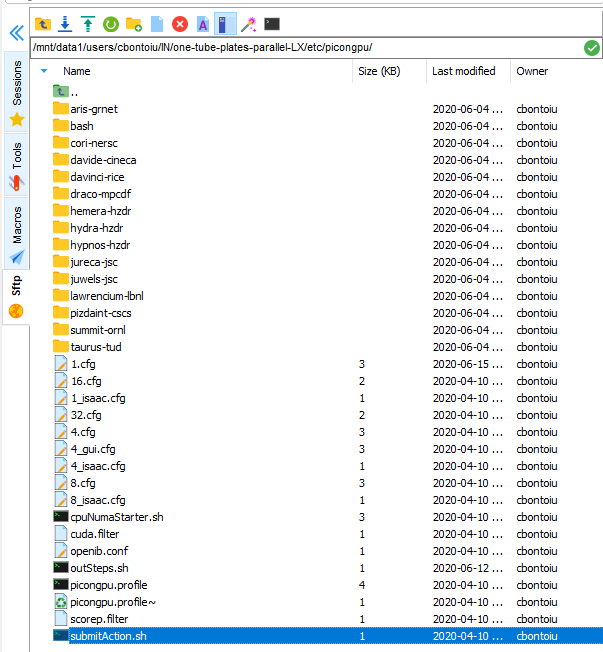
Do you mean that I should create a file called submit.start? But where to place it and how to include it in the complete syntax for submitting the job? So far I have used:
tbg -s sbatch -c etc/picongpu/1.cfg -t etc/picongpu/hemera-hzdr/gpu.tpl ../../OUT/test
cbontoiu
on 16 Jun 2020
The file submit.start should be in the tbg/ directory of your simulation directory (which you send as zip file a while ago). From the content of that zip file, I would guess you will find the submit.start file in
/users/cbontoiu/volatile/OUT/testG/tbg/submit.start
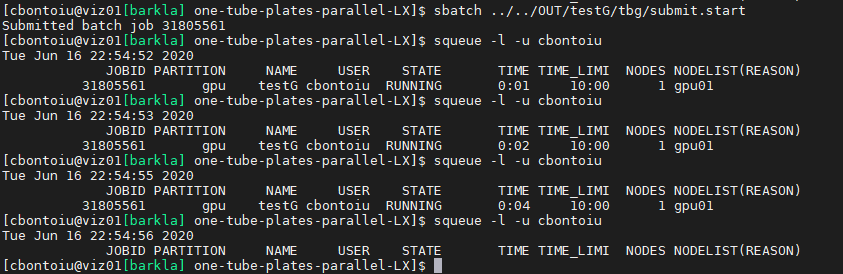
I tried with
and obtained this error
no binary 'cuda_memtest' available or compute node is not exclusively allocated, skip GPU memory test
the runtime was about 4 seconds.
I will try with a typical LWFA case from picongpu distribution. maybe it is something wrong with my model, though I understand that the chances are very low.
cbontoiu
on 16 Jun 2020
I have new error message in the stderr file coming from the LWFA example which I submitted as tbg -s sbatch -c etc/picongpu/1.cfg -t etc/picongpu/hemera-hzdr/gpu.tpl ../../OUT/test_C.
no binary 'cuda_memtest' available or compute node is not exclusively allocated, skip GPU memory test /tmp/slurmd/job31805592/slurm_script: line 75: 1529 Illegal instruction mpiexec /users/cbontoiu/volatile/OUT/test_C/input/bin/picongpu -d 1 1 1 -g 192 1024 12 -s 2048 --periodic 0 0 1
line 75 is here
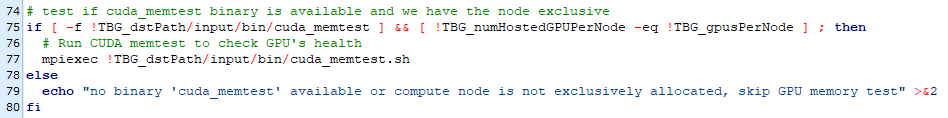
and it appears that the cuda_memtest.sh cannot be executed.
I then submitted directly the sumbit.start file from the ouput/tbg folder of the initial launch, and ontained the same message:
no binary 'cuda_memtest' available or compute node is not exclusively allocated, skip GPU memory test /tmp/slurmd/job31805595/slurm_script: line 75: 22938 Illegal instruction mpiexec /users/cbontoiu/volatile/OUT/test_C/input/bin/picongpu -d 1 1 1 -g 192 1024 12 -s 2048 --periodic 0 0 1
cbontoiu
on 17 Jun 2020
@cbontoiu Strange. Great you tried the default LWFA example.
The used etc/picongpu/hemera-hzdr/gpu.tpl is similar enough to your Barkla setup (except for memory allocation), that it should cause no error.
Line 75 refers to your submit.start as posted here (and thus to the end of the if clause surrounding your picongpu call) and not your *.tpl file as shown here. Thus this is the place an error should occur with picongpu.
A assume you used #SLURM --exclusive again. Could you please add a line in you submit.start before mpiexec ..../picongu that contains nvidia-smi - just to check that there are no other processes on the GPU - and resubmit that job?
What does your simOutput/ directory contain? What else is in your stdout and stderr?
PrometheusPi
on 17 Jun 2020
Had an offline chat with @PrometheusPi . So there are a few things we are not sure or confused about. Perhaps it would be easier to have a video call or at least some form of instant messages, to quickly try things out and see the outcome. Are you up for it @cbontoiu ? I don't think that would be a long session.
sbastrakov
on 17 Jun 2020
Had an offline chat with @PrometheusPi . So there are a few things we are not sure or confused about. Perhaps it would be easier to have a video call or at least some form of instant messages, to quickly try things out and see the outcome. Are you up for it @cbontoiu ? I don't think that would be a long session.
@sbastrakov and @PrometheusPi Great idea. And I could give you access to my computer via TeamViewer. But I just woke up. Maybe we can arrange this meeting for 10 o'clock UK time, 11 o'clock in Germany? Could you set up a zoom conference? If not I can try, though I never did it so far. Thank you, Regards
cbontoiu
on 17 Jun 2020
Helllo @cbontoiu , yes, I am also not available right now. Let's continue discussing the call details, links etc. by email, I will use one from your .tpl file.
sbastrakov
on 17 Jun 2020
A quick thing to try out, could be before the call or during it:
In the end of your .tpl file, replace line
mpiexec !TBG_dstPath/input/bin/picongpu !TBG_author !TBG_programParams
with
mpirun -npernode !TBG_gpusPerNode -n !TBG_tasks !TBG_dstPath/input/bin/picongpu !TBG_author !TBG_programParams
sbastrakov
on 17 Jun 2020
@sbastrakov It appears that the #SLURM --exclusive command was not accepted so far and I submitted again the job with #SBATCH --exclusive. I can see that my job is pending on the gpu partition now.
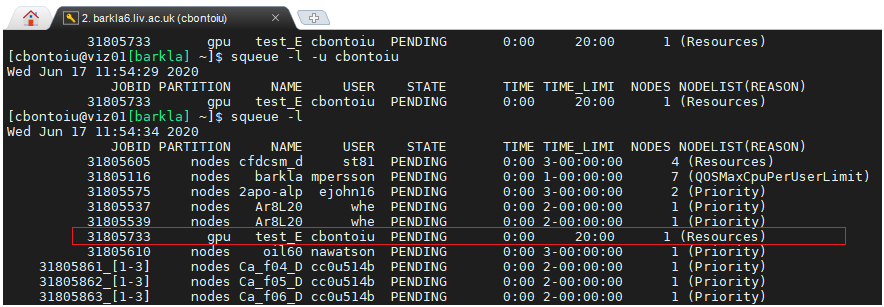
However, at this time of the day the cluster is busy, my job is waiting already for an hour or so, and I wonder if wouldn't be a better idea to meet later in the afternoon/evening to be able to submit and get results. Of course it depends on your agenda. I am available anytime.
cbontoiu
on 17 Jun 2020
(edit : this was found out during the VC)
By default mpiexec cannot find the number of processes and one has to set
mpiexec -n !TBG_tasks !TBG_dstPath/input/bin/picongpu !TBG_author !TBG_programParams
instead of
mpiexec !TBG_dstPath/input/bin/picongpu !TBG_author !TBG_programParams
However, after doing that picongpu seems no to see any gpu even though we set ```
SBATCH --gres=gpu:!TBG_gpusPerNodeat the top of the.tplfile. Thus we try to add the coomandnvidia-smi```
cbontoiu
on 17 Jun 2020
Working hypothesis: MPI from spack does not interact well with SLURM on the cluster. And so the GPUs allocated, and other parameters like number of processes are not properly passed through. @cbontoiu will try to tell spack to use the installed MPI instead of building its own. I guess like that
sbastrakov
on 17 Jun 2020
I included a packages.yaml file in the .spack folder as I know it works from my experience with a single machine. The file looks like
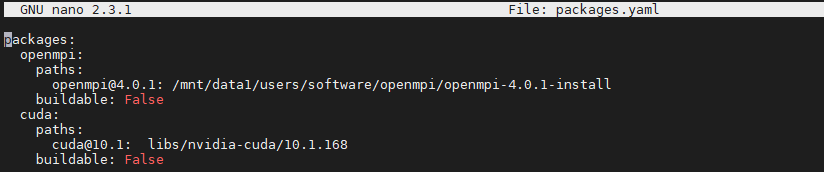
and I am not sure about the path to CUDA so I asked the admin. But there is a difference from what I have done in the past. If before CUDA and openMPI were never installed by Spack because they had existed on the system as indicated by packages.yaml, now we are in the situation in which CUDA and openMPI are installed by Spack and I am not sure if the packages.yaml file helps. Maybe we need to uninstall those two from Spack?
The slurm error message is now

cbontoiu
on 18 Jun 2020
I think you need to spack install another version of PIConGPU, that would use that CUDA and MPI packages. I assume, now performing source $HOME/.../spack/share/spack/setup-env.sh would activate them. And running spack install afterwards will install PIConGPU using those. I think it makes sense not to delete the previous installation, but just use another name.
Also I think your path for the CUDA package should be absolute, starting with /.
sbastrakov
on 18 Jun 2020
@sbastrakov
Uninstalling/installing picongpu with the packages.yaml defined as above didn't make any change to my problem.
I ended up wiping out my Spack installation and reinstalling Spack and then picongpu with a different packages.yaml file because it seems that picongpu cannot work with openmpi version 4 and they have either this or version 1 which is a bit too old I think.
The error was
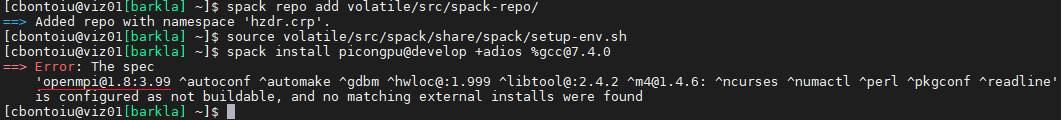
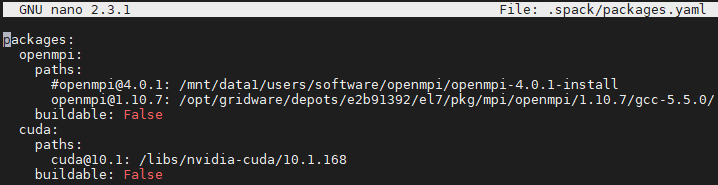
The installation went smoothly and I can confirm that there is no openmpi nor cuda folder in spack/opt/ which means that cluster-wide modules were accepted.
However, now picongpu cannot compile the laser-wakefield model. It gives the error below.
[ 76%] Building CXX object CMakeFiles/cupla.dir/users/cbontoiu/volatile/src/spack/opt/spack/linux-centos7-skylake_avx512/gcc-7.4.0/picongpu-develop-vgawfgqgvs2elxkxj4it6ctr7yk5hriu/thirdParty/cupla/src/event.cpp.o
In file included from /users/cbontoiu/volatile/src/spack/opt/spack/linux-centos7-skylake_avx512/gcc-7.4.0/picongpu-develop-vgawfgqgvs2elxkxj4it6ctr7yk5hriu/thirdParty/cupla/include/cupla/namespace.hpp:23:0,
from /users/cbontoiu/volatile/src/spack/opt/spack/linux-centos7-skylake_avx512/gcc-7.4.0/picongpu-develop-vgawfgqgvs2elxkxj4it6ctr7yk5hriu/thirdParty/cupla/src/stream.cpp:22:
/users/cbontoiu/volatile/src/spack/opt/spack/linux-centos7-skylake_avx512/gcc-7.4.0/picongpu-develop-vgawfgqgvs2elxkxj4it6ctr7yk5hriu/thirdParty/cupla/include/cupla/defines.hpp:81:6: error: #error "there is no accelerator selected, please run `ccmake .` and select one"
#error "there is no accelerator selected, please run `ccmake .` and select one"
^~~~~
Maybe you can help me with this?
cbontoiu
on 18 Jun 2020
@cbontoiu Could you please check whether in your profile, the is a PIC_BACKEND defindes as e.g. export PIC_BACKEND="cuda:60" and whether is is exportet correctly, by running echo $PIC_BACKEND?
If you go into your build directory .build and run ccmake ., what backend is set there?
PrometheusPi
on 19 Jun 2020
@PrometheusPi
The succession of commands is
source src/spack/share/spack/setup-env.sh
spack load picongpu@develop +adios %[email protected] && export PIC_BACKEND="cuda:70"
ccmake .
I get this output
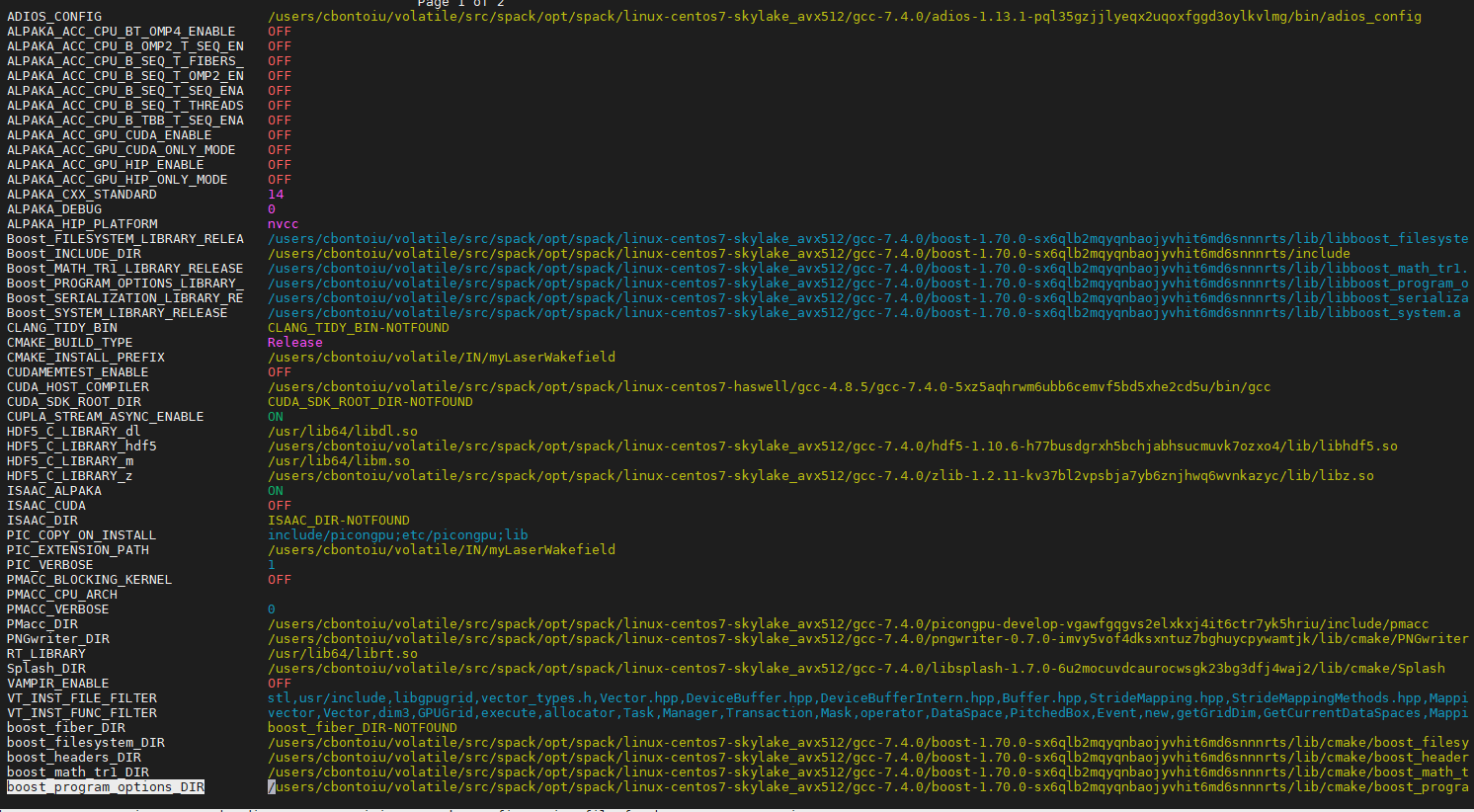

cbontoiu
on 19 Jun 2020
Hello @cbontoiu . So from the last output there are still no backends enabled. I think the issue may be that export PIC_BACKEND="cuda:70" should be done before source src/spack/share/spack/setup-env.sh, or somehow inside it: if that file sources some more profiles, perhaps there.
sbastrakov
on 19 Jun 2020
Hello @sbastrakov Here are my commands in a new terminal without spack/setup-end.sh being loaded by default.
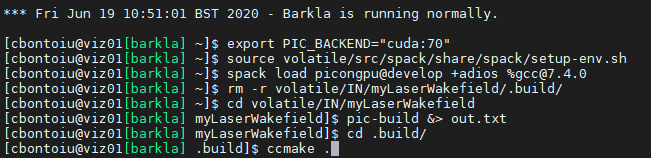
The error is the same and the output of ccmake . is the same.
cbontoiu
on 19 Jun 2020
Not sure why this happens. Perhaps some details are in the out.txt generated, could you attach it?
sbastrakov
on 19 Jun 2020
Here it is. Thank you
cbontoiu
on 19 Jun 2020
Thanks. This file had the important details indeed. So it seems pic-build has the correct options to build PIConGPU with CUDA. However, cmake could not find your CUDA installation (line 32 of the attached file), and thus no backend got enabled which caused the later error of "there is no accelerator selected".
Why was CUDA not found: I think it's worth checking your spack configuration. Also you may try to just load it with module load before.
sbastrakov
on 19 Jun 2020
@sbastrakov
I think the answer relies on this message from the admin
"The modules mpi/openmpi/4.0.1/gcc-5.5.0 and libs/nvidia-cuda/10.1.168/bin are certainly correctly defined and working well. It depends on how the yaml file interprets these paths (root path, or library path or binary path etc). The path /libs/nvidia-cuda/10.1.168 is wrong, you could find the path info by “module show libs/nvidia-cuda/10.1.168/bin” .
The advantage of Spack is that it can automatically install any dependency packages if there are not available. As you mentioned, it may be compatibility issue of OpenMPI. It might be better to let Spack do it automatically rather than specify them manually (as it is easy to get wrong).
With the build script /mnt/data1/users/software/Spack/build_PIConGPU_gcc7.3.0_auto.sh, openmpi 3.1.5 is automatically installed (see /mnt/data1/users/software/Spack/build_gcc7.3.0_auto.log). "
It give me this
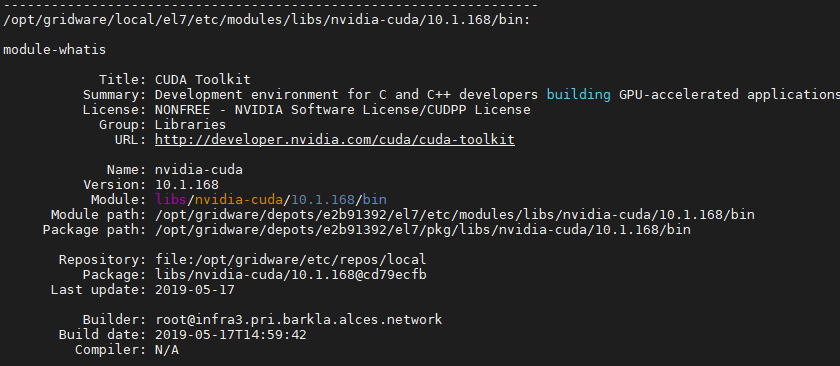
so I will update my packages.yaml
An important question is if picongpu can work with openmpi 4.
cbontoiu
on 19 Jun 2020
About openmpi 4: PIConGPU should work with it. In case it does not, we are not aware of it yet, and will fix it
sbastrakov
on 19 Jun 2020
I asked because yesterday I had the error message shown below
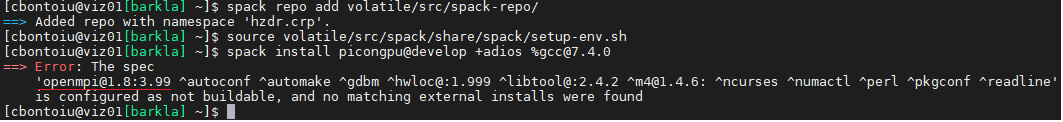
so now I work with openmpi 1. They don't seem to have other versions installed.
cbontoiu
on 19 Jun 2020
@cbontoiu Did using openMPI 1 worked for you?
PrometheusPi
on 22 Jun 2020
@cbontoiu Did using openMPI 1 worked for you?
@PrometheusPi
It didn't work but I will try again today with the admins. Obviously, I couldn't find a correct path for openmpi on the cluster. I will update you as soon as I complete everything I can do myself. Thank you.
cbontoiu
on 22 Jun 2020
@sbastrakov
Hello,
Does the end of my cfg file look right?
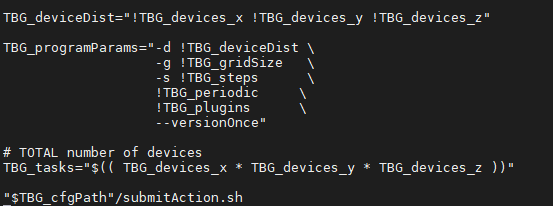
shouldn't there be an equal sign after TBG_cfgPath inside the quotation marks?
cbontoiu
on 24 Jun 2020
Hello @cbontoiu . No, the attached piece seems good, there should not be = in that fragment.
sbastrakov
on 24 Jun 2020
Hello @sbastrakov and @PrometheusPi
I still have no solution to my problem, though Spack is now installed with external openmpi 4 (I released the limitation in packages.py) and CUDA 10. The tpl file was adjusted by the admins.
Please suggest any further options that I can try.
Kind Regards,
Cristian
cbontoiu
on 26 Jun 2020
I have an update from Barkla admins:
It appears the tool template batch generator (tbg) doesn’t behave as expected. I guess the workflow of tbg might be:
- Generate job script file according to configuration files (.cfg) and job template files (.tpl).
- Submit the job script to the job scheduler.
The tbg has generated job script (tbg/submit.start, looks ok), but seems to completely ignore all the Slurm options:
#SBATCH --partition=gpu
#SBATCH --time=0:5:00
#SBATCH --job-name=TZ
#SBATCH --nodes=1
#SBATCH --ntasks=6
#SBATCH --gres=gpu:1
#SBATCH --mail-type=NONE
#SBATCH [email protected]
#SBATCH -D /users/cbontoiu/volatile/OUT/TZ
#SBATCH -o stdout
#SBATCH -e stderr
So everything will use the default settings (for example, 8 hours rather than 5 minutes, default partition rather than GPU partition). You have also seen the CUDA errors because it ran on a non-GPU node.
In addition, mpi/openmpi/4.0.1 was built with GCC 5.5.0. So there might be some computability issue.
I would suggest to use the OpenMPI installed by Spack.
In order to track the problem, I would suggest:
Check the previously generated job script (tbg/submit.start), and manually submit this job script to the job scheduler.
If this works, then we know there might be some problem for tbg in submitting jobs. If this doesn’t work properly, execute the commands in tbg/submit.start interactively on viz01 or viz02 (don’t use Slurm job scheduler).
Then you will see where it fails if there is any.
cbontoiu
on 26 Jun 2020
@cbontoiu Could you please share the full tpl, cfg file and the combined submit.start. Could be that #!/usr/bin/env bash is missing in the tpl file.
You can also try to submit the submit.start file directly with sbatch submit.start tbg is calling internally the same command.
 psychocoderHPC
on 26 Jun 2020
psychocoderHPC
on 26 Jun 2020
@cbontoiu @sbastrakov showed me the submit.start you used. The slurm docu is saying that you are not allowed to have any command before #SBATCH
https://slurm.schedmd.com/sbatch.html
The batch script may contain options preceded with "#SBATCH" before any executable commands in the script.
sbatch will stop processing further #SBATCH directives once the first non-comment non-whitespace line has been
reached in the script.
sbatch exits immediately after the script is successfully transferred to the Slurm controller and assigned
a Slurm job ID.
So @cbontoiu I think you should try to edit your .tpl file so that it starts with
#!/bin/bash -l (or another bash), followed by all the #SBATCH lines and then all the other stuff. Just reordering of lines. Then the submit.start is generated using the same structure as .tpl and so should also have this new ordering.
sbastrakov
on 26 Jun 2020
There is some difference and the job is submitted differently as you can check in the image below
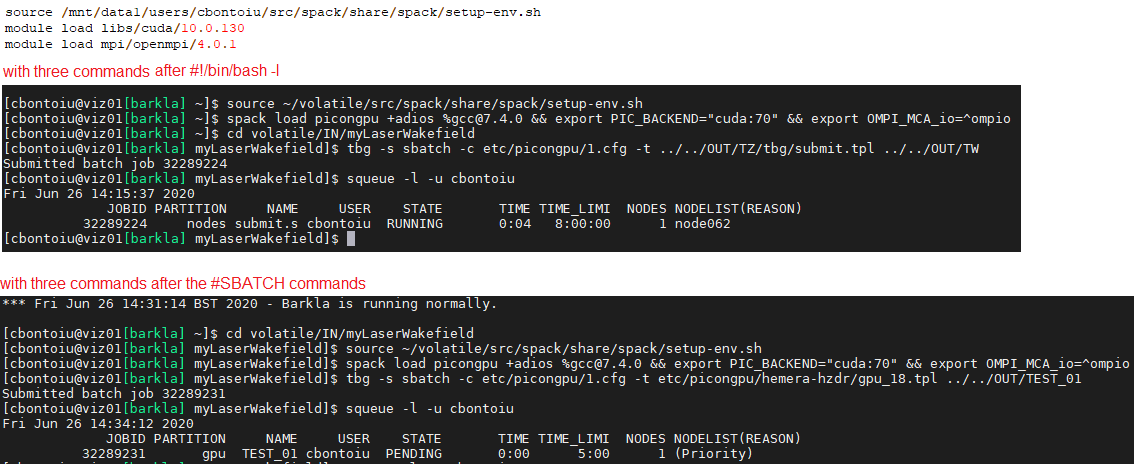
I also attach my new tpl file which is a fusion of what you indicated and what advices I got from the admin.
Thank you. I will come back with the update once the job gets a chace to run.
cbontoiu
on 26 Jun 2020
Looks like good news: now (in the second version) it actually tries to run on the GPU partition and with 5 minutes.
sbastrakov
on 26 Jun 2020
Here are the indications I got yesterday from the Barkla cluster admins, for the tpl file
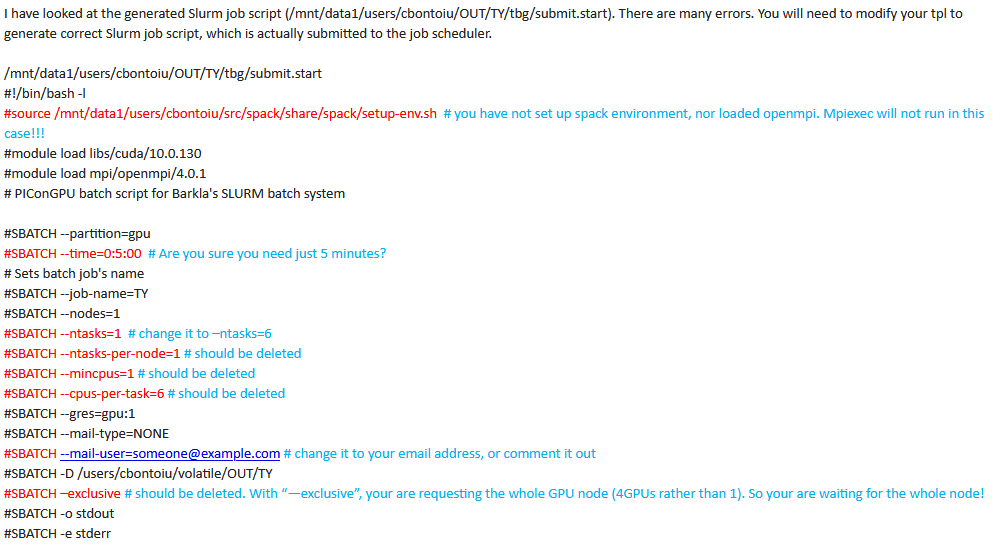
cbontoiu
on 26 Jun 2020
Okay, so the source ... probably should be there, just after all the #SBATCH stuff.
With tasks, ntasks-per-node, mincpus, cpus-per-task there are normally multiple ways to set it all up, but cluster may have some specifics. So I guess it's reasonable to follow admin's suggestions.
The mail-user was already set up properly (with your actual email) in your earlier files. Not sure why did it get changed back to the default.
--exclusive yes, in general case it should not be used there. Again, your earlier attached files did not have it there.
sbastrakov
on 26 Jun 2020
Okay, so the
source ...probably should be there, just after all the#SBATCHstuff.
Withtasks, ntasks-per-node, mincpus, cpus-per-taskthere are normally multiple ways to set it all up, but cluster may have some specifics. So I guess it's reasonable to follow admin's suggestions.
Themail-userwas already set up properly (with your actual email) in your earlier files. Not sure why did it get changed back to the default.
--exclusiveyes, in general case it should not be used there. Again, your earlier attached files did not have it there.
Yes, the gpu_18 - Copy_tpl.txt file attached above, is a copy of what I have now as the best version. If it fails again, I am going to reinstall PIConGPU this time with openmpi and cuda inside Spack and then try again.
cbontoiu
on 26 Jun 2020
OK, so my job got a chance to run and this time there was no messsage from slurm but other two files appeared.
stderr
no binary 'cuda_memtest' available or compute node is not exclusively allocated, skip GPU memory test
Primary job terminated normally, but 1 process returned
a non-zero exit code. Per user-direction, the job has been aborted.
mpiexec noticed that process rank 0 with PID 22211 on node gpu01 exited on signal 4 (Illegal instruction).
and stdout
Running program...
I will not try to submit with sbatch submit.start
cbontoiu
on 26 Jun 2020
I think this "illegal instruction" may be caused by compiler versions or flags mismatch (e.g. the node architecture does not match the target architecture used to compile the code)
sbastrakov
on 26 Jun 2020
I think this "illegal instruction" may be caused by compiler versions or flags mismatch (e.g. the node architecture does not match the target architecture used to compile the code)
Yes, obviously there is an incompatibility as you can see from this picture. I think openmpi and cuda are used along the first route while picongpu runs along the second route. Because the latest picongpu@develop moved to [email protected] I am forced to use spack install picongpu@develop +adios %[email protected]
I also have this outcome
cbontoiu
on 26 Jun 2020
So maybe our original suggestion about MPI (during the call) was not correct, and the issue was only in the # SBATCH commands being in the wrong place (after source ...). Did you try to not use system openMPI by removing it from the .yml file and so build one from spack?
sbastrakov
on 29 Jun 2020
Well, I wanted to have a fresh install of Spack and I ran into other troubles like in the image below. Surprisingly the command spack compiler list | grep -q [email protected] || spack install [email protected] && spack load [email protected] && spack compiler add failed this time, so I had no chance to install picongpu

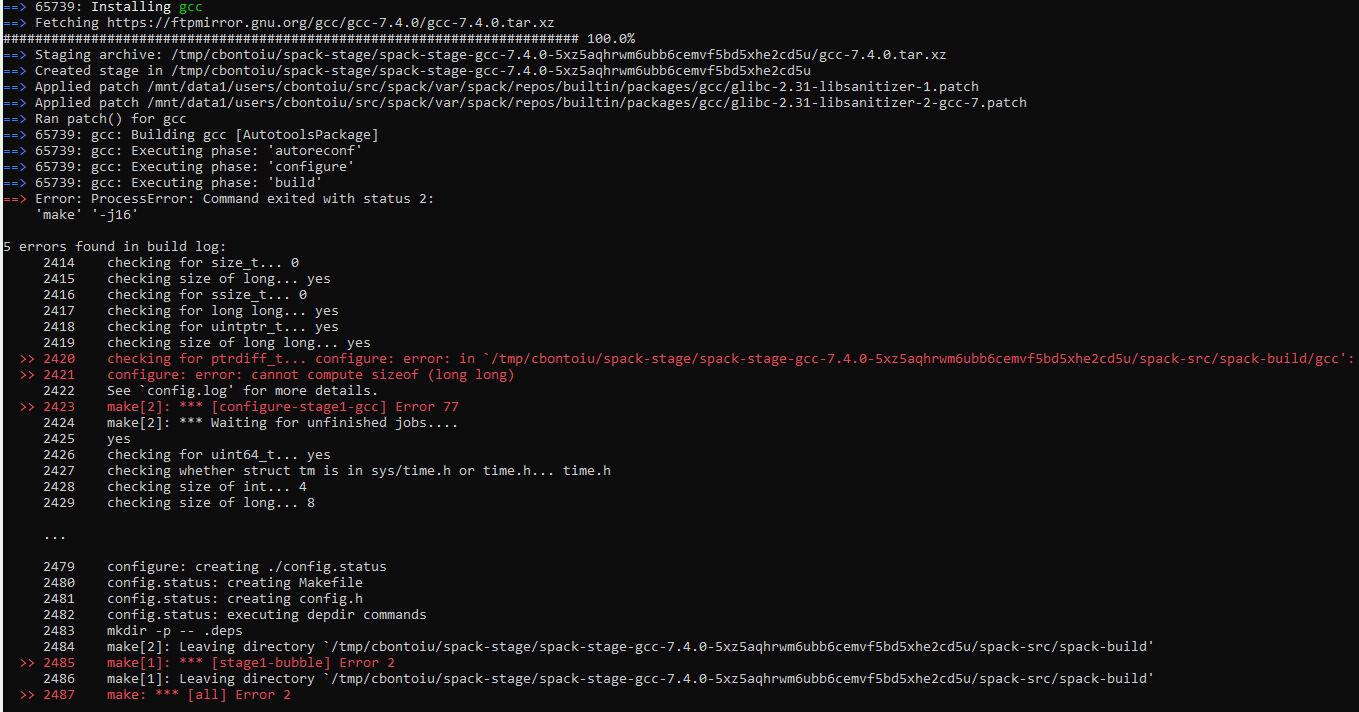
cbontoiu
on 29 Jun 2020
So you mean that previously that command worked? And the version is 7.4.0, not 7.3.0?
sbastrakov
on 29 Jun 2020
So you mean that previously that command worked? And the version is 7.4.0, not 7.3.0?
Yes, previosuly version 7.4.0 was installed and I managed to compile a picongpu model. As you know this is required to work with the latest picongpu@develop. But now, something happened, either with the cluster or with the Spack distribution so I started an issue https://github.com/spack/spack/issues/17294 and also wrote to the cluster admin.
cbontoiu
on 29 Jun 2020
A suggestion from our colleague @SimeonEhrig about the "illegal instruction" error before. It could be that the head node has Skylake architecture and so when you compile on it, including installing spack packages, it compiles for this architecture, but the GPU nodes have older Haswell CPUs. We could check if that's true by looking at node hardware specification (it seems to not be present on the web page, which is the only thing available from the outside). To go around it you might try to spack install and compile everything on a GPU node
Edit: to try this out, could also still build on the head node but add target=x86_64 at the end of the spack install commands.
sbastrakov
on 29 Jun 2020
Concerning the gcc 7.4.0 requirement I am honestly a little surprised about it, and would not think this is the case.
sbastrakov
on 29 Jun 2020
Concerning the gcc 7.4.0 requirement I am honestly a little surprised about it, and would not think this is the case.
Hello. This explanation about a possible incompatibility appeared as part of a reply for an older issue #3247
home/cristi/PIC_INPUT/one-tube-plates-parallel-LX/include/alpaka/elem/Traits.hpp(36):
error #135: namespace "std" has no member "remove_volatile_t"
This could be an issue because the develop branch switched to C++14 with #3242
but the spack recipe is not tested for it.
cbontoiu
on 29 Jun 2020
I understand that C++14 is required for the develop branch. However, I believe all gcc versions from 6.1 support it.
sbastrakov
on 30 Jun 2020
Since GCC 6.1, C++14 is default: https://gcc.gnu.org/git/?p=gcc.git;a=commitdiff;h=0801f419440c14f6772b28f763ad7d40f7f7a580
 SimeonEhrig
on 30 Jun 2020
SimeonEhrig
on 30 Jun 2020
@sbastrakov Hello, we made some progress on the cluster. The admin installed Spack for me with its own openmpi and cuda. I ontained the error atached below, running the file tbg/submit.start line by line in the terminal.
and then the admin suggested :
......................
The error “CUDA error: invalid device ordinal” can be eliminated by setting up the CUDA_VISIBLE_DEVICES environment variable (see https://stackoverflow.com/questions/39649102/how-do-i-select-which-gpu-to-run-a-job-on), for example. export CUDA_VISIBLE_DEVICES=1 The error “CUDA error: no kernel image is available for execution on” is normally caused by incompatibility of different versions of CUDA. In addition, if cuda_memtest source code is hard-coded to use the first GPU (CUDA_VISIBLE_DEVICES=0), the code might report error when large memory is needed as the first GPU is already used by other programs (“nvidia-smi” to check the GPU info).
...................
Applying export CUDA_VISIBLE_DEVICES=1 and again running the file tbg/submit.start line by line in the terminal I see that for the first time the cuda memory test started but there are still two errors in the simOutput folder attached below.
cuda_memtest_viz01.pri.barkla.alces.network_31.err.txt
cuda_memtest_viz01.pri.barkla.alces.network_27.err.txt
These errors refer to the PIConGPU distribution for Spack. Is there anything that we can do about, please?
Regards and thanks,
Cristian
cbontoiu
on 7 Jul 2020
Hello @cbontoiu .
I am not sure what goes wrong with cuda_memtest. Perhaps @psychocoderHPC has an idea?
Does the same error occur in PIConGPU as well? You can disable running cuda_memtest (it's just a small separate program) by commenting out the (second) line
# Run CUDA memtest to check GPU's health
mpiexec !TBG_dstPath/input/bin/cuda_memtest.sh
in your .tpl file.
sbastrakov
on 7 Jul 2020
@sbastrakov @psychocoderHPC
I tried running again the commands in tbg/submit.start skiping the cuda memory test and obtained the error below. It complains about zlib_1.2.9 but I have zlib_1.1.11 installed through Spack. Should I release the limitation in packages.py and reinstall picongpu@develop with Spack?
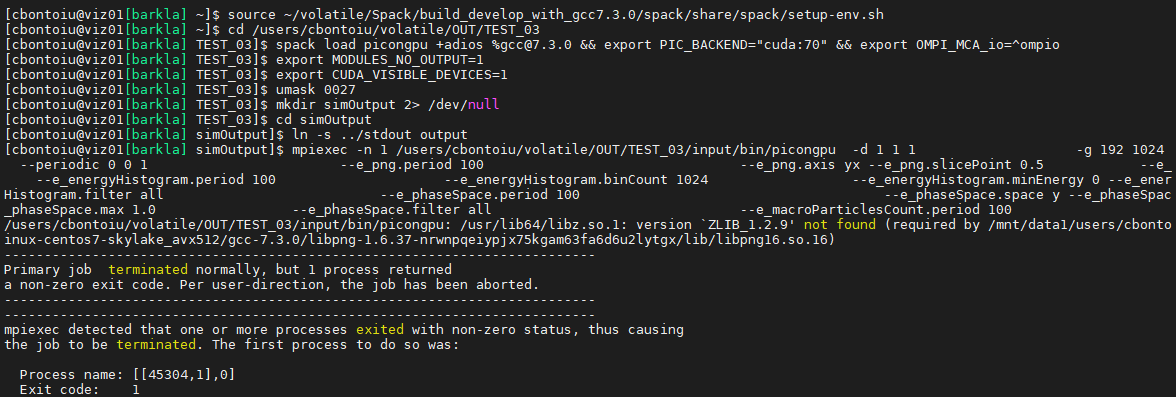
cbontoiu
on 7 Jul 2020
Hello @cbontoiu . So this zlib_1.2.9 requirement comes from libpng. I am honestly not sure why this isn't handled by Spack, but it basically means you need to have 1.2.9 at least and so would need to raise the requirement.
sbastrakov
on 8 Jul 2020
@sbastrakov Do you mean that I need to reinstall picongpu@develop with zlib 1.2.9 instead of zlib 1.2.11 as it is now? And I can do this downgrading the version in the /spack-repo/packages/picongpu/packages.py
cbontoiu
on 9 Jul 2020
Sorry, I am a little confused. In the previous message, you told that you have version 1.1.11 installed (not 1.2.11). Having 1.2.11 should be no problem.
sbastrakov
on 9 Jul 2020
Sorry, I am a little confused. In the previous message, you told that you have version 1.1.11 installed (not 1.2.11). Having 1.2.11 should be no problem.
It was a mistake from my side. Indeed Spack installed version 1.2.11, so the question is if downgrading will be fine.
cbontoiu
on 9 Jul 2020
I am honestly not sure. It looks like the 1.2.9 requirement comes from somewhere else. Is it possible to install the other version in addition to 1.2.11 ? Or check if the system has a module for it.
sbastrakov
on 9 Jul 2020
@sbastrakov @psychocoderHPC
I tried running again the commands in
tbg/submit.startskiping the cuda memory test and obtained the error below. It complains aboutzlib_1.2.9but I havezlib_1.1.11installed through Spack. Should I release the limitation inpackages.pyand reinstallpicongpu@developwithSpack?
@cbontoiu could you please post (as text file not image) the output of the following commands:
# run all commands from your image until `ln -s ../stdout output`
# run now
env
@psychocoderHPC @sbastrakov
[cbontoiu@viz01[barkla] ~]$ cd volatile/OUT/TEST_03
[cbontoiu@viz01[barkla] TEST_03]$ spack load picongpu +adios %[email protected] && export PIC_BACKEND="cuda:70" && export OMPI_MCA_io=^ompio
[cbontoiu@viz01[barkla] TEST_03]$ export MODULES_NO_OUTPUT=1
[cbontoiu@viz01[barkla] TEST_03]$ export CUDA_VISIBLE_DEVICES=1
[cbontoiu@viz01[barkla] TEST_03]$ umask 0027
[cbontoiu@viz01[barkla] TEST_03]$ mkdir simOutput 2> /dev/null
[cbontoiu@viz01[barkla] TEST_03]$ cd simOutput
[cbontoiu@viz01[barkla] simOutput]$ run now
bash: run: command not found...
[cbontoiu@viz01[barkla] simOutput]$ mpiexec -n 1 /users/cbontoiu/volatile/OUT/TEST_03/input/bin/picongpu -d 1 1 1 -g 192 1024 12 -s 2048 --periodic 0 0 1 --e_png.period 100 --e_png.axis yx --e_png.slicePoint 0.5 --e_png.folder pngElectronsYX --e_energyHistogram.period 100 --e_energyHistogram.binCount 1024 --e_energyHistogram.minEnergy 0 --e_energyHistogram.maxEnergy 1000 --e_energyHistogram.filter all --e_phaseSpace.period 100 --e_phaseSpace.space y --e_phaseSpace.momentum py --e_phaseSpace.min -1.0 --e_phaseSpace.max 1.0 --e_phaseSpace.filter all --e_macroParticlesCount.period 100 --versionOnce
/users/cbontoiu/volatile/OUT/TEST_03/input/bin/picongpu: /usr/lib64/libz.so.1: version `ZLIB_1.2.9' not found (required by /mnt/data1/users/cbontoiu/Spack/build_develop_with_gcc7.3.0/spack/opt/spack/linux-centos7-skylake_avx512/gcc-7.3.0/libpng-1.6.37-nrwnpqeiypjx75kgam63fa6d6u2lytgx/lib/libpng16.so.16)
--------------------------------------------------------------------------
Primary job terminated normally, but 1 process returned
a non-zero exit code. Per user-direction, the job has been aborted.
--------------------------------------------------------------------------
--------------------------------------------------------------------------
mpiexec detected that one or more processes exited with non-zero status, thus causing
the job to be terminated. The first process to do so was:
Process name: [[31088,1],0]
Exit code: 1
--------------------------------------------------------------------------
[cbontoiu@viz01[barkla] simOutput]$
sry there is maybe some misunderstanding please execute the following commands:
cd volatile/OUT/TEST_03
spack load picongpu +adios %[email protected] && export PIC_BACKEND="cuda:70" && export OMPI_MCA_io=^ompio
export MODULES_NO_OUTPUT=1
export CUDA_VISIBLE_DEVICES=1
umask 0027
mkdir simOutput 2> /dev/null
cd simOutput
env
@psychocoderHPC Here it is. Thank you.
cbontoiu
on 10 Jul 2020
Thanks could you please run:
cd volatile/OUT/TEST_03
spack load picongpu +adios %[email protected] && export PIC_BACKEND="cuda:70" && export OMPI_MCA_io=^ompio
export MODULES_NO_OUTPUT=1
export CUDA_VISIBLE_DEVICES=1
umask 0027
mkdir simOutput 2> /dev/null
cd simOutput
ldd /users/cbontoiu/volatile/OUT/TEST_03/input/bin/picongpu
echo "-------------------------------------"
mpiexec -n 1 ldd /users/cbontoiu/volatile/OUT/TEST_03/input/bin/picongpu
@psychocoderHPC I must say thanks to you instead.
Here are the errors. I collected them sepparately for ldd and mpiexec
cbontoiu
on 10 Jul 2020
@psychocoderHPC Hello again. Is there any hint about what should I do?
cbontoiu
on 15 Jul 2020
Could could try to install zlib with spack install zlib
could you try
cd volatile/OUT/TEST_03
spack load picongpu +adios %[email protected] && export PIC_BACKEND="cuda:70" && export OMPI_MCA_io=^ompio
export MODULES_NO_OUTPUT=1
export CUDA_VISIBLE_DEVICES=1
umask 0027
mkdir simOutput 2> /dev/null
cd simOutput
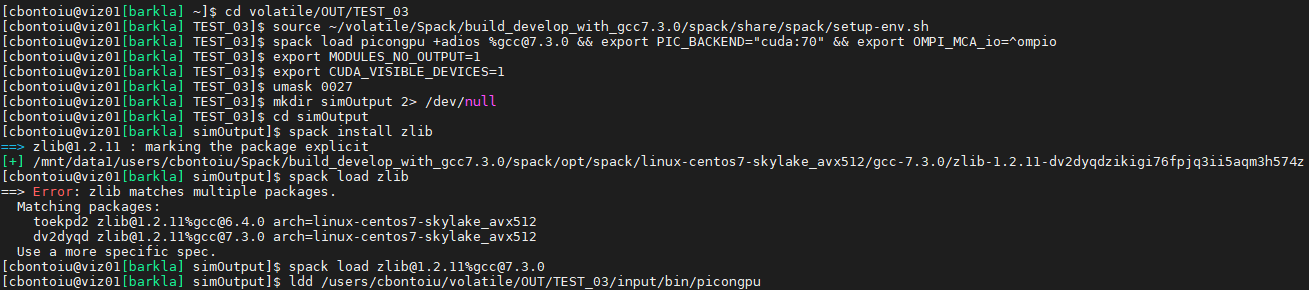
spack install zlib
spack load zlib
ldd /users/cbontoiu/volatile/OUT/TEST_03/input/bin/picongpu
For some reason the logs show that the system provided zlib is used.
psychocoderHPC
on 15 Jul 2020
@psychocoderHPC Thank you. These were my commands
and the output of ldd is attached below:
cbontoiu
on 15 Jul 2020
sry but you need to run it again, the output said that you need to specify the zlib package you like to load because you have already build two different versions.
cd volatile/OUT/TEST_03
spack load picongpu +adios %[email protected] && export PIC_BACKEND="cuda:70" && export OMPI_MCA_io=^ompio
export MODULES_NO_OUTPUT=1
export CUDA_VISIBLE_DEVICES=1
umask 0027
mkdir simOutput 2> /dev/null
cd simOutput
spack install zlib
spack load [email protected]%gcc7.3.0
ldd /users/cbontoiu/volatile/OUT/TEST_03/input/bin/picongpu
Hello @psychocoderHPC,
Attached you will find all my commands and the error output from the terminal. Still the problem is that ZLIB 1.2.9 is missing.
cbontoiu
on 17 Jul 2020
I am not sure why the loaded spack libz is not found. Let us check if the files are available.
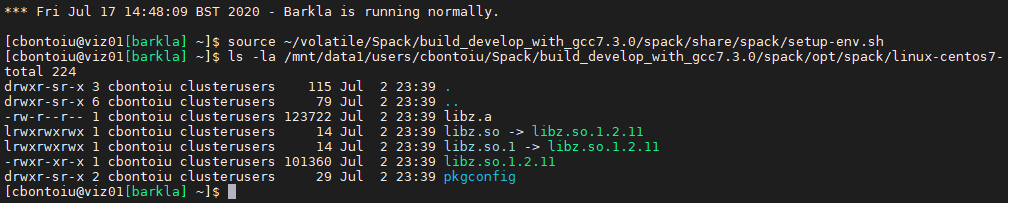
spack load picongpu +adios %[email protected] && export PIC_BACKEND="cuda:70" && export OMPI_MCA_io=^ompio
ls -la /mnt/data1/users/cbontoiu/Spack/build_develop_with_gcc7.3.0/spack/opt/spack/linux-centos7-skylake_avx512/gcc-7.3.0/zlib-1.2.11-dv2dyqdzikigi76fpjq3ii5aqm3h574z/lib
@psychocoderHPC Here it is
cbontoiu
on 17 Jul 2020
ok hopefully last try:
cd volatile/OUT/TEST_03
spack load picongpu +adios %[email protected] && export PIC_BACKEND="cuda:70" && export OMPI_MCA_io=^ompio
export MODULES_NO_OUTPUT=1
export CUDA_VISIBLE_DEVICES=1
umask 0027
mkdir simOutput 2> /dev/null
cd simOutput
export LD_LIBRARY_PATH=/mnt/data1/users/cbontoiu/Spack/build_develop_with_gcc7.3.0/spack/opt/spack/linux-centos7-skylake_avx512/gcc-7.3.0/zlib-1.2.11-dv2dyqdzikigi76fpjq3ii5aqm3h574z/lib:$LD_LIBRARY_PATH
ldd /users/cbontoiu/volatile/OUT/TEST_03/input/bin/picongpu
If the ldd from the command above still points to the libz under /usr/local please compile the picnogpu example again and provide all output, including a ldd to the newly compiled PIConGPU binary.
psychocoderHPC
on 17 Jul 2020
@psychocoderHPC
OK, I compiled again the model and the output of the compilation process only is here
I don't know where should I point the ldd command now.
cbontoiu
on 17 Jul 2020
@cbontoiu you pass the binary of PIConGPU to ldd
ldd <path_to_the_input_set>/bin/picongpu
<path_to_the_input_set> must be substituted with the path where you called pic-build, it should be ldd ~/volatile/IN/myLaserWakefield/bin/picongpu
psychocoderHPC
on 21 Jul 2020
@psychocoderHPC Thank you.
Here is the output of the ldd command called in the bin/picongpu folder
ldd_output_new_compile.txt
cbontoiu
on 21 Jul 2020
@ax3l Do you have any idea what could be the problem that libz is used from the system and not from the loaded spack modules?
We checked already the environment . LD_LIBRARY_PATH looks good.
@cbontoiu Currently I am running out of ideas.
psychocoderHPC
on 21 Jul 2020
@psychocoderHPC Maybe I should ask the admin to install picongpu system wide with dependencies installed on the cluster, avoiding thus Spack?
cbontoiu
on 23 Jul 2020
or maybe someone could help me with access to a cluster where picongpu runs during the summer holiday, please?
cbontoiu
on 23 Jul 2020
Hello @cbontoiu . I think this may work. When installing manually, these docs might be useful, it has the command line flags we ourselves use when installing packages manually. Of course, it can be that some of those libraries are already available on the cluster, in this case we load them in the .profile files, like those
sbastrakov
on 24 Jul 2020
Hello again on this topic @psychocoderHPC and @sbastrakov
Barkla admin at Liverpool tries to get PIConGPU running on the cluster and he came back with the following message shown below. Given your experience on other clusters, could you help with a suggestion please? Thank you. Cristian
.......................
From the information you provided, picongpu can generate a working executable if there is a right version of system zlib library (>=1.2.11). However, there is some problem to link the spack-installed zlib library or an external zlib library. This might be a bug in picongpu configuration or we haven’t found the hidden option (at least I have not found it yet).
We will need to ask the developers about how to link the spack-installed or external zlib library (rather than system zlib library in /usr/lib64/ or /lib/x86_64-linux-gnu/) when generating the picongpu executable. Could you raise this issue to the developers?
I could try building picongpu from source, but I suspect there might be the same issue with the zlib library.
..........................
cbontoiu
on 19 Aug 2020
Hello @cbontoiu
picongpu can generate a working executable if there is a right version of system zlib library (>=1.2.11)
This is correct, zlib with at least that version is a mandatory dependency of PIConGPU
I think the easiest way to provide a correct zlib installation for PIConGPU build is to follow this scheme, the last 3 (second-level) bullet points. I believe this can be applied for both spack and installing from source.
For installing from source, we ourselves used it on Piz Daint supercomputer as part of the profile there. First, we set ZLIB_ROOT to the actual installation we want to use (in that case, the manually installed by us) at line 53, then we extend other environment variables at lines 61 and 71. Note that we prepend rather than append, so that this version is found first: if there were also a visible another (e.g. system-wide) installation that does not work for us, prepending would work, but not appending.
For spack, I think one can do almost the same. Just do these 3 lines in the terminal after doing all the usual environment setup and just before running pic-build. This should result in effectively the same environment state but with correct zlib when the build is starting.
sbastrakov
on 19 Aug 2020
Related issues
cbontoiu
·
3Comments
sbastrakov
·
3Comments
 ax3l
·
4Comments
ax3l
·
4Comments
 hightower8083
·
4Comments
hightower8083
·
4Comments
 saipavankalyan
·
3Comments
saipavankalyan
·
3Comments
Most helpful comment
@sbastrakov Hello, we made some progress on the cluster. The admin installed Spack for me with its own openmpi and cuda. I ontained the error atached below, running the file
tbg/submit.startline by line in the terminal.latest_error.txt
and then the admin suggested :
......................
The error “CUDA error: invalid device ordinal” can be eliminated by setting up the CUDA_VISIBLE_DEVICES environment variable (see https://stackoverflow.com/questions/39649102/how-do-i-select-which-gpu-to-run-a-job-on), for example. export CUDA_VISIBLE_DEVICES=1 The error “CUDA error: no kernel image is available for execution on” is normally caused by incompatibility of different versions of CUDA. In addition, if cuda_memtest source code is hard-coded to use the first GPU (CUDA_VISIBLE_DEVICES=0), the code might report error when large memory is needed as the first GPU is already used by other programs (“nvidia-smi” to check the GPU info).
...................
Applying
export CUDA_VISIBLE_DEVICES=1and again running the filetbg/submit.startline by line in the terminal I see that for the first time the cuda memory test started but there are still two errors in thesimOutputfolder attached below.cuda_memtest_viz01.pri.barkla.alces.network_31.err.txt
cuda_memtest_viz01.pri.barkla.alces.network_27.err.txt
These errors refer to the
PIConGPUdistribution forSpack. Is there anything that we can do about, please?Regards and thanks,
Cristian