Tskit: Bug in map_mutations() for polytomy
@jeromekelleher
I used map_mutations to remap genotypes onto my inferred tree sequence and got unexpected results. The total number of mutations was _higher_ than that inferred by tsinfer. I whittled it down to a reproducible, minimal example:
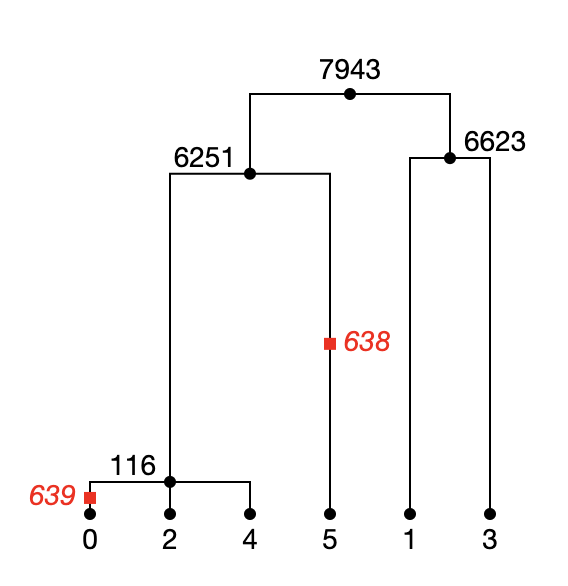
Here is a tree in the (simplified down) tree sequence. The genotype array is:
[1, 0, 0, 0, 0, 1] and alleles are ('C', 'T').
Now we run:
tree.map_mutations(np.array([1, 0, 0, 0, 0, 1]), ("C", "T"))
And we get _three_ mutations:
('C', [{'id': -1, 'site': -1, 'node': 6251, 'time': nan, 'derived_state': 'T', 'parent': -1, 'metadata': b''}, {'id': -1, 'site': -1, 'node': 2, 'time': nan, 'derived_state': 'C', 'parent': 0, 'metadata': b''}, {'id': -1, 'site': -1, 'node': 4, 'time': nan, 'derived_state': 'C', 'parent': 0, 'metadata': b''}])
Am I correct in thinking this is a bug? Doc of map_mutations() states:
The current implementation uses the Fitch parsimony algorithm to determine the minimum number of state transitions required to explain the data. In this model, transitions between any of the non-missing states is equally likely.
But map_mutations infers an extra mutation. Perhaps could be an issue with polytomies?
Here's code to reproduce:
import tskit
import io
def polytomy_tree_ts():
nodes = io.StringIO("""\
id is_sample time
0 1 0
1 1 0
2 1 0
3 1 0
4 1 0
5 1 0
6 0 1
7 0 2
8 0 2
9 0 3
""")
edges = io.StringIO("""\
left right parent child
0 1 6 0,2,4
0 1 7 6,5
0 1 8 1,3
0 1 9 7,8
""")
sites = io.StringIO("""\
position ancestral_state
0.5 0
""")
mutations = io.StringIO("""\
site node derived_state
0 0 1
0 5 1
""")
return tskit.load_text(nodes=nodes, edges=edges,sites=sites, mutations=mutations, strict=False)
ex = polytomy_tree_ts()
ex.first().map_mutations(var.genotypes, ("G", "C"))
 awohns
awohns
All 7 comments
Very weird... I think it must be a bug, and yes, something to do with the polytomy. I can't see how the second one is anything bug less parsimonious.
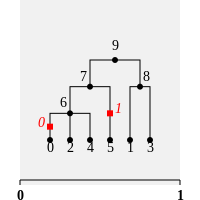

import tskit
import io
def polytomy_tree_ts():
nodes = io.StringIO(
"""\
id is_sample time
0 1 0
1 1 0
2 1 0
3 1 0
4 1 0
5 1 0
6 0 1
7 0 2
8 0 2
9 0 3
"""
)
edges = io.StringIO(
"""\
left right parent child
0 1 6 0,2,4
0 1 7 6,5
0 1 8 1,3
0 1 9 7,8
"""
)
sites = io.StringIO(
"""\
position ancestral_state
0.5 0
"""
)
mutations = io.StringIO(
"""\
site node derived_state
0 0 1
0 5 1
"""
)
return tskit.load_text(
nodes=nodes, edges=edges, sites=sites, mutations=mutations, strict=False
)
ex = polytomy_tree_ts()
ancestral_state, mutations = ex.first().map_mutations([1, 0, 0, 0, 0, 1], ("G", "C"))
ex.draw_svg("tmp1.svg")
tables = ex.dump_tables()
tables.sites.clear()
tables.mutations.clear()
tables.sites.add_row(position=0.5, ancestral_state=ancestral_state)
for mutation in mutations:
tables.mutations.add_row(
site=0, derived_state=mutation.derived_state, node=mutation.node
)
tables.compute_mutation_parents()
ts = tables.tree_sequence()
ts.draw_svg("tmp2.svg")
 jeromekelleher
on 11 Nov 2020
jeromekelleher
on 11 Nov 2020
Well tracked down, thanks @awohns!
jeromekelleher
on 11 Nov 2020
Well, this was a major oversight on my part - the Fitch algorithm that we're using is only for binary trees. It seems that we get a reconstruction that works with non-binary trees, but that's not parsimonious, as in this case. It never occured to me that the algorithm would be binary-only, but I clearly didn't do my research properly and didn't actually test on non-binary trees either.
I'll implement a more general algorithm and report back.
@benjeffery - I think it's best we put the 0.3.3 release on hold for this, as this code is used in tsinfer and we want to ship a fix asap.
jeromekelleher
on 19 Nov 2020
Also, FYI @hyanwong and @awohns this affects tsinfer from a performance perspective because internally it uses Fitch parsimony to compress the likelihood values on the tree. So, we're not compressing the likelihoods as well as we might, which will affect performance (some good news, I guess).
jeromekelleher
on 19 Nov 2020
Well found @jeromekelleher - thanks for following up on this. It does seem like a very important one to fix.
 hyanwong
on 19 Nov 2020
hyanwong
on 19 Nov 2020
Yep we'll hold off on 0.3.3 for this for sure. No rush as it is a self-imposed deadline.
 benjeffery
on 19 Nov 2020
benjeffery
on 19 Nov 2020
Looks like I need to implement Hartigan's algorithm, which isn't all that different to Fitch. It's pretty heavily optimised low-level code doing bit-set manipulation, though, so we'll see.
jeromekelleher
on 19 Nov 2020
Related issues
 petrelharp
·
8Comments
petrelharp
·
8Comments
 daniel-goldstein
·
8Comments
jeromekelleher
·
3Comments
hyanwong
·
3Comments
jeromekelleher
·
9Comments
daniel-goldstein
·
8Comments
jeromekelleher
·
3Comments
hyanwong
·
3Comments
jeromekelleher
·
9Comments
Most helpful comment
Well tracked down, thanks @awohns!