Currently we have no way of recording the precise age of a mutation. All that we know is that it occured somewhere on a branch. Most of the time this isn't an issue, but there are situtations in which we need to record this precise time (see, https://github.com/tskit-dev/msprime/pull/946, e.g.).
We can add a MutationTable.time column easily enough. If the mutation time is not specified (the default) then we set it to the time of the mutation's node. If multiple mutations are added on the same branch without times being specified, then they will have the same time (oh well, no big deal I guess).
In terms of the file format, we make this optional in the same way that Edge metadata is optional (it would be simplest to call set_columns to implement this though). It might be best to do #506 first, therefore.
As this is backward compatible, we don't need to increment the file version number.
We will break some C code since mutation_table_add_row et al will need a new parameter, which most naturally comes between the current parent and derived_state. I guess providing TSK_NULL indicates you want the default for this. Since we're already breaking some C code, this seems fine.
@benjeffery, any thoughts here?
 jeromekelleher
jeromekelleher
All 32 comments
Supporting information that this is a good idea: SLiM already records the time, but in metadata.
 petrelharp
on 6 Apr 2020
petrelharp
on 6 Apr 2020
fwdpy11 does the same thing. If you keep tables in the native (fwdpp) format, the mutation's origin time is with respect to time moving forwards. If you dump to the tskit format, then origin times are changed to be pastwards from the last time point simulated.
 molpopgen
on 6 Apr 2020
molpopgen
on 6 Apr 2020
I'm reassured we all think this is a good idea!
jeromekelleher
on 7 Apr 2020
Do we know why SLiM includes this in metadata? It seems to be a core enough property of a mutation to warrant inclusion as a column.
I agree on getting #506 done first and making this backwards compatible. This will be the first "non-trivial" default value so care will need to be taken in documenting the behaviour.
 benjeffery
on 7 Apr 2020
benjeffery
on 7 Apr 2020
Do we know why SLiM includes this in metadata? It seems to be a core enough property of a mutation to warrant inclusion as a column.
It was just because it wasn't implemented in tskit and we decided there were other things to be getting on with, I'd imagine.
jeromekelleher
on 7 Apr 2020
A though: once this is implemented, we should have sort_tables sort mutations by time, which satisfies the current requirement that parents come before children, but would make the ordering produced by sort_tables less ambiguous (not totally, if in discrete time, though). Perhaps we should make "sorted by time" a requirement of the mutation table, also.
petrelharp
on 26 May 2020
I've started working on this. Above it says that if TSK_NULL is passed as the time to tsk_mutation_table_add_row then it should default to the node time. However I don't see how tsk_mutation_table_add_row has access to the node time as it only has the node id.
benjeffery
on 3 Jun 2020
Just had a discussion with @jeromekelleher and @awohns on this issue.
Any way of loading an old TS that doesn't have the mutation time column seems complex and a kludge. The mutation loading code would need to have the node table and do lots of look ups. It is also not clear how tsk_mutation_table_add_row could provide a default.
It seems a cleaner path to make mutation time a mandatory column (bumping the major file version). Then tskit upgrade would add mutation times by spreading them equally across each edge in a one-off procedure. Mutation time would be mandatory in add_row. @petrelharp thoughts?
benjeffery
on 3 Jun 2020
I agree - making mutation time mandatory sounds like the right way to go. Hm: it seems like we'll really want to get msprime and SLiM producing mutation times right away, to avoid a period of awkwardness? This will be easy to add in SLiM, but would require a release to get adoption. I guess what I'm saying is that a lot of people might be encountering this.
About spreading the times out over the branch - also, agree! At first I was nervous about adding extra fake information, but we're not using mutation times currently, so I think it's good.
Hm - it occurs to me that some methods - eg tsinfer? - don't have any natural way to assign times along a branch, so it could be useful to allow clients to ignore times in tables (eg leave them all NULL), and provide a method to assign times uniformly along a branch, much like compute_mutation_parents and deduplicate_sites? Since we need to write this method anyhow? (to be clear, I think the time column should make sense for a valid tree sequence, I'm talking about 'just a collection of tables')
This will be great to get in there for the future "time slice" statistics!
petrelharp
on 3 Jun 2020
Hm - it occurs to me that some methods - eg tsinfer? - don't have any natural way to assign times along a branch, so it could be useful to allow clients to ignore times in tables (eg leave them all NULL), and provide a method to assign times uniformly along a branch, much like compute_mutation_parents and deduplicate_sites? Since we need to write this method anyhow? (to be clear, I think the time column should make sense for a valid tree sequence, I'm talking about 'just a collection of tables')
Great idea @petrelharp, let's do this.
jeromekelleher
on 3 Jun 2020
I've done most of the work to put mutation time through the code. I've also added checks so that for a valid tree sequence times need to be not null, greater or equal to the node time and less than or equal to the parent mutation if there is one. I think I need to add less than or equal to the node time above, right if no parent? That means following the edge which means a lookup in the edge table.
I'm now working on the method to put times on existing, timeless mutations, which is needed to get existing tests working so has to be in the same PR. I think I need to check integrity (with time checking turned off) before adding times, right? Or for efficiency do we assume a valid tree sequence is passed to these kinds of methods?
benjeffery
on 4 Jun 2020
I think I need to add less than or equal to the node time above, right if no parent? That means following the edge which means a lookup in the edge table.
That's a good idea, but do we have the ID of the edge that a mutation falls on? I don't think we can do this unless we do (But - maybe we should have the ID of the edge in the mutation table - it's actually more useful than just the child node. That's a big change though.)
I think I need to check integrity (with time checking turned off) before adding times, right? Or for efficiency do we assume a valid tree sequence is passed to these kinds of methods?
Definitely we want to check the integrity first. Efficiency is always a secondary consideration to safety/correctness unless a C API user tells us not to check. Put another way, the default behaviour should always be to do any checks that are necessary to guarantee safety.
jeromekelleher
on 4 Jun 2020
I'm currently looking at how to get a mutation->edge mapping from the current data model, I need this not only for the consistency check as mentioned above, but also when creating fake times for legacy tree sequences that don't have them. Here's the part of the current data model that's relevant:
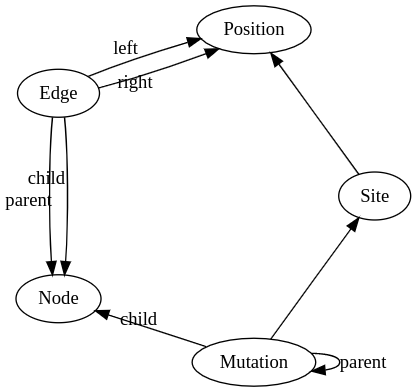
Prototyping in python this seemed the most obvious way:
mutations_above_node = defaultdict(list)
for mut_idx, mut in enumerate(mutations):
mutations_above_node[mut.node].append((mut_idx, mut))
mutation_edges = [-1] * len(mutations)
for edge_idx, edge in enumerate(edges):
for mut_idx, mut in mutations_above_node[edge.child]:
if edge.left <= sites[mut.site].position < edge.right:
mutation_edges[mut_idx] = edge_idx
To translate this to C I'd need to figure out the best structure for the mutations_above_node ragged array. An Iliffe vector would be the obvious way, but not very cache friendly.
Another way that I dismissed would be something like:
for mut_idx, mut in enumerate(tables.mutations):
overlapping_edges = find_edges_for_site(tables.sites[mut.site])
for edge_idx, edge in overlapping_edges:
if edge.child == mut.child:
mutation_edges[mut_idx] = edge_idx
Where find_edges_for_site is based on some kind of interval tree - this doesn't seem a good option though as I assume num_edges > num_mutations and building such a tree would be O(n log n) in the number of edges.
Maybe I'm missing something here as @jeromekelleher mentioned making a table of edges in one pass...
benjeffery
on 5 Jun 2020
Why not just build the trees and iterate along the sequence that way? See e.g. compute_mutation_parents. Seems like any other way of doing it - if mutations aren't associated to edges - will be just as complex.
petrelharp
on 5 Jun 2020
I think building the trees is the simplest way all right, following the example of compute_mutaton_parents.
jeromekelleher
on 5 Jun 2020
Thanks both, now I'm thinking with tree sequences!
One more question as I'm afraid. For a given edge with mutations, we're planning to assign times by spreading the mutations across the edge. I just spoke to @jeromekelleher who pointed out that the sets of mutations that get spread should be _per site_. So if there are two mutations on an edge but at different sites, they both get put at the mid-time of the edge.
If that is the case then I don't think I need to worry about finding out which mutations share an edge as two mutations that have the same site and node necessarily are on the same edge, correct??
EDIT: Of course I still need to find the edge to get the length! :facepalm:
benjeffery
on 5 Jun 2020
If that is the case then I don't think I need to worry about finding out which mutations share an edge as two mutations that have the same site and node necessarily are on the same edge, correct??
That's true. They'll probably be adjacent in the table, but that's probably not guaranteed, so you will need to go through all the mutations for a site and group them by node.
jeromekelleher
on 5 Jun 2020
Or, gee, you could just assign times lazily to each mutation but then go back and adjust previously-assigned times one the same branch, by checking if the mutation.parent has the same node, since they are guaranteed to come in order going down the tree.
petrelharp
on 5 Jun 2020
Oh - ps - is the idea to assign them exactly uniformly, or randomly uniformly? I guess I vote for the latter - more useful, anyhow.
petrelharp
on 5 Jun 2020
Currently doing exactly uniformly (i.e. a single mutation gets put on the edge mid-time). But easily adjust.
benjeffery
on 5 Jun 2020
Well, if it was randomly uniformly and there was a public method to re-compute it, then that would be very useful for averaging over mutation placement on the branches!
petrelharp
on 5 Jun 2020
Oh - ps - is the idea to assign them exactly uniformly, or randomly uniformly? I guess I vote for the latter - more useful, anyhow.
I would be very much in favour of exactly uniformly! Where do we get the randomness from, otherwise? We can't use GSL, libc's RNGs are rubbish, and there's the seeding headache to worry about. This would greatly complicate things.
jeromekelleher
on 5 Jun 2020
Might be worth making a Python version of this and opening a PR @benjeffery? We'll need it for testing anyway, and that way we can feed in concretely.
jeromekelleher
on 5 Jun 2020
Oh - ps - is the idea to assign them exactly uniformly, or randomly uniformly? I guess I vote for the latter - more useful, anyhow.
I would be very much in favour of exactly uniformly! Where do we get the randomness from, otherwise? We can't use GSL, libc's RNGs are rubbish, and there's the seeding headache to worry about. This would greatly complicate things.
I guess a callback receiving the two node times and returning a double could allow external libs to generate randomness?
molpopgen
on 5 Jun 2020
I would be very much in favour of exactly uniformly! Where do we get the randomness from, otherwise? We can't use GSL, libc's RNGs are rubbish, and there's the seeding headache to worry about. This would greatly complicate things.
Darn, because averaging over uniform random mutation placement is something that I want to do.
petrelharp
on 5 Jun 2020
This library has a compatible licence but doesn't directly do random double https://www.pcg-random.org/using-pcg-c.html. We'd need to do something like they do here http://mumble.net/~campbell/tmp/random_real.c, but that has no licence.
benjeffery
on 5 Jun 2020
Alternatively, if there was just a method to obtain the edge, or parent node time, then this randomization would be easy enough to do in pytohn.
petrelharp
on 5 Jun 2020
This is a lot of extra complexity to take on now, and I feel like we'll get bogged down in it. How about this: we add an extra argument called random which if not NULL should be an array of at least num_mutations doubles between 0 and 1. Then, if random is specified we use one of those random values per mutation to put it in the right place. We could work out the details later, but for now only do the non-random case.
Callbacks etc are a pain, and don't scale when working with Python.
jeromekelleher
on 5 Jun 2020
Alternatively, if there was just a method to obtain the edge, or parent node time, then this randomization would be easy enough to do in pytohn.
Yes, that's better! If we know the edge then we could do this in Python with the tables API. Probably wouldn't be as fast as the approach above though, if you wanted to do it many times.
jeromekelleher
on 5 Jun 2020
Probably wouldn't be as fast as the approach above though, if you wanted to do it many times.
It'd just be some numpy vector addition/multiplication and a call to set_columns?
petrelharp
on 5 Jun 2020
It'd just be some numpy vector addition/multiplication and a call to set_columns?
It's not that easy if you have multiple mutations on an edge though, is it? You'd end up having to loop over the sites, I'd have thought.
jeromekelleher
on 5 Jun 2020
I like the "passing doubles in" approach. I'll implement that.
benjeffery
on 5 Jun 2020
Related issues
 hyanwong
·
7Comments
hyanwong
·
3Comments
hyanwong
·
7Comments
hyanwong
·
3Comments
 mufernando
·
6Comments
mufernando
·
6Comments
 bhaller
·
5Comments
petrelharp
·
6Comments
bhaller
·
5Comments
petrelharp
·
6Comments
Most helpful comment
This is a lot of extra complexity to take on now, and I feel like we'll get bogged down in it. How about this: we add an extra argument called
randomwhich if not NULL should be an array of at leastnum_mutationsdoubles between 0 and 1. Then, if random is specified we use one of those random values per mutation to put it in the right place. We could work out the details later, but for now only do the non-random case.Callbacks etc are a pain, and don't scale when working with Python.