Rdkit: EnumerateStereisomers has inconsistent behavior
Configuration:
- RDKit Version: 2020.03.01
- Operating system: Windows 10
- Python version (if relevant): 3.6.10
- Are you using conda?: Yes
- If you are using conda, which channel did you install the rdkit from? rdkit
Description:
I notice that Chem.EnumerateStereoisomers gives different number of isomers when presented with input molecules that are "identical". I noticed this behavior for some in-memory mol objects. In order to make it reproducible I encoded an example mol object as JSON to be able to share here. The structure was downloaded from PubChem and has one stereocenter -- thus I expect 2 entries in the enumeration. I only observe 1. If I export and re-import from MolBlock format, I get the expected 2 isomers.
print(rdkit.__version__)
enumsi_opt = EnumerateStereoisomers.StereoEnumerationOptions(maxIsomers=20, onlyUnassigned=False, tryEmbedding = True)
mol = Chem.rdMolInterchange.JSONToMols("""{"commonchem":{"version":10},"defaults":{"atom":{"z":6,"impHs":0,"chg":0,"nRad":0,"isotope":0,"stereo":"unspecified"},"bond":{"bo":1,"stereo":"unspecified"}},"molecules":[{"atoms":[{"z":8},{"z":8},{"z":7},{"z":7},{"z":7},{"z":7},{"z":7},{"z":7,"impHs":2},{"impHs":1,"stereo":"ccw"},{"impHs":2},{"impHs":2},{"impHs":2},{"impHs":2},{},{},{},{},{},{},{"impHs":1},{"impHs":1},{"impHs":1},{"impHs":1},{"impHs":2},{"impHs":1},{"impHs":1},{},{},{"impHs":1},{"impHs":1},{"impHs":1},{"impHs":1},{"impHs":1}],"bonds":[{"bo":2,"atoms":[0,15]},{"atoms":[1,26]},{"atoms":[1,27]},{"atoms":[2,10]},{"atoms":[2,12]},{"atoms":[2,15]},{"atoms":[3,4]},{"atoms":[8,3]},{"atoms":[3,13]},{"bo":2,"atoms":[4,16]},{"bo":2,"atoms":[5,13]},{"atoms":[5,20]},{"atoms":[6,17]},{"bo":2,"atoms":[6,20]},{"atoms":[7,17]},{"atoms":[8,9]},{"atoms":[8,10]},{"atoms":[9,11]},{"atoms":[11,12]},{"atoms":[13,14]},{"atoms":[14,16]},{"bo":2,"atoms":[14,17]},{"atoms":[15,19]},{"atoms":[16,18]},{"bo":2,"atoms":[18,21]},{"atoms":[18,22]},{"bo":2,"atoms":[19,23]},{"atoms":[21,24]},{"bo":2,"atoms":[22,25]},{"bo":2,"atoms":[24,26]},{"atoms":[25,26]},{"bo":2,"atoms":[27,28]},{"atoms":[27,29]},{"atoms":[28,30]},{"bo":2,"atoms":[29,31]},{"bo":2,"atoms":[30,32]},{"atoms":[31,32]}],"conformers":[{"dim":2,"coords":[[11.8866,-4.8319],[8.8814,5.6391],[9.4172,-5.6391],[7.0174,-2.4781],[7.8928,-1.2711],[4.2989,-2.7711],[3.0,-0.521],[4.2989,1.7288],[7.4833,-3.904],[6.4816,-5.0205],[8.9511,-4.2133],[6.9477,-6.4463],[8.4154,-6.7555],[5.598,-2.0211],[5.598,-0.521],[10.885,-5.9484],[7.0174,-0.064],[4.2989,0.2289],[7.4833,1.3617],[11.3509,-7.3741],[3.0,-2.0211],[6.4816,2.4781],[8.9511,1.6711],[12.8187,-7.6835],[6.9477,3.904],[9.4172,3.0968],[8.4154,4.2133],[10.3492,5.9484],[11.3509,4.8319],[10.8151,7.3741],[12.8187,5.1413],[12.2829,7.6835],[13.2847,6.5672]]}],"extensions":[{"name":"rdkitRepresentation","formatVersion":1,"toolkitVersion":"2020.03.1","aromaticAtoms":[3,4,5,6,13,14,16,17,18,20,21,22,24,25,26,27,28,29,30,31,32],"aromaticBonds":[6,8,9,10,11,12,13,19,20,21,24,25,27,28,29,30,31,32,33,34,35,36],"atomRings":[[2,12,11,9,8,10],[4,3,13,14,16],[5,20,6,17,14,13],[21,24,26,25,22,18],[28,30,32,31,29,27]]}]}]}""")[0]
stereoisomers = list(Chem.EnumerateStereoisomers.EnumerateStereoisomers(mol, enumsi_opt))
print(len(stereoisomers))
mol = Chem.MolFromMolBlock(Chem.MolToMolBlock(mol))
stereoisomers = list(Chem.EnumerateStereoisomers.EnumerateStereoisomers(mol, enumsi_opt))
print(len(stereoisomers))
Output:
2020.03.01
1
2
 paconius
paconius
All 4 comments
confirmed
 greglandrum
on 8 Jun 2020
greglandrum
on 8 Jun 2020
This is happening because molecules constructed from JSON do not have the atomic properties set which indicate which atoms are possible chiral centers.
We should think about modifying EnumerateStereoisomers() to always call AssignStereochemistry() (if it's already been called the function will return early, so it's no additional work).
In the meantime, the workaround is:
In [24]: Chem.AssignStereochemistry(mol,force=True,flagPossibleStereoCenters=True)
In [26]: len(list(EnumerateStereoisomers.EnumerateStereoisomers(mol,enumsi_opt)))
Out[26]: 2
Chem.AssignStereochemistry(mol,force=True,flagPossibleStereoCenters=True)
In [26]: len(list(EnumerateStereoisomers.EnumerateStereoisomers(mol,enumsi_opt)))
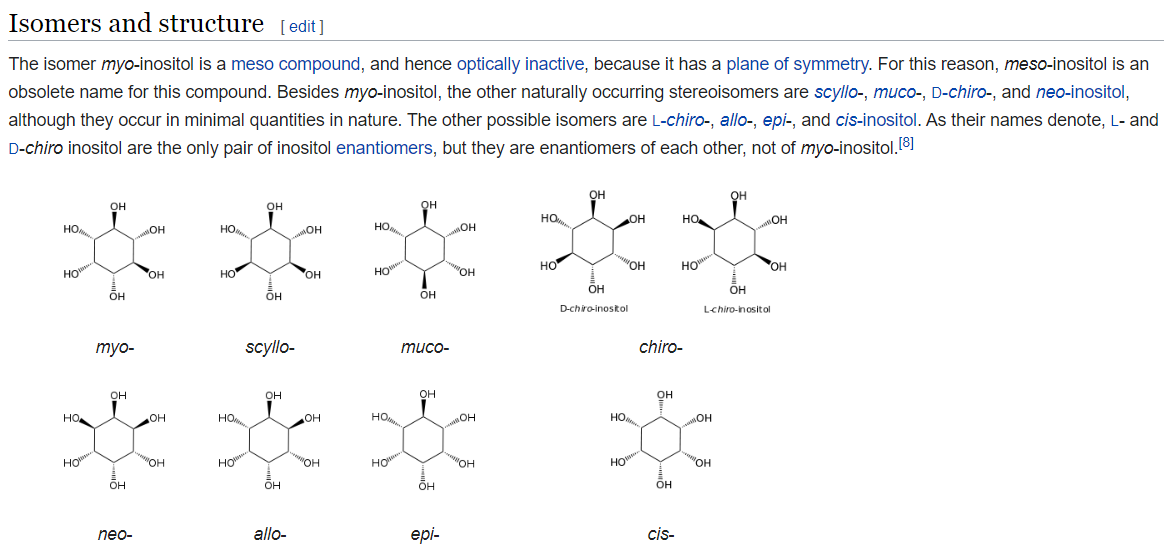
Thanks Greg. This seems to fix the issue for the molecule included above. However I notice it doesn't fix the issue for a compound like inositol (see below). Perhaps there is another layer to this?
from rdkit import Chem
from rdkit.Chem import AllChem
from rdkit.Chem import EnumerateStereoisomers
inositol = Chem.MolFromSmiles('C1(C(C(C(C(C1O)O)O)O)O)O')
Chem.AssignStereochemistry(inositol,force=True,flagPossibleStereoCenters=True)
enumsi_opt = EnumerateStereoisomers.StereoEnumerationOptions(maxIsomers=20, onlyUnassigned=False, tryEmbedding = True)
len(list(EnumerateStereoisomers.EnumerateStereoisomers(inositol,enumsi_opt)))
Output:
1
For the expected isomers of inositol see wikipedia:
paconius
on 8 Jun 2020
The problem with inositol is a different one: the code that identifies possible stereocenters does not currently work for unspecified para (dependent) stereocenters like what you have here.
I'm working on fixing that, but it's a slow process. Hopefully we'll have something ready for the next release.
greglandrum
on 10 Jun 2020
Related issues
 mc-robinson
·
5Comments
mc-robinson
·
5Comments
 hjuinj
·
6Comments
hjuinj
·
6Comments
 BillLawrence111
·
3Comments
BillLawrence111
·
3Comments
 jaimergp
·
3Comments
jaimergp
·
3Comments
 asmunder
·
3Comments
asmunder
·
3Comments