Openff-toolkit: Issue creating smirnoff systems with example script
I have been trying to create Smirnoff systems using a few different molecules to no avail.
I am using the example code from the toluene notebook, and have altered it for my test cases.
# Create an OpenFF Topology of ligand from a pdb file.
ligand_pdbfile = PDBFile('test.pdb')
ligand = Molecule.from_smiles('CCCCOc1ccccc1')
#ligand = Molecule.from_file('test.sdf')
print(ligand_pdbfile.topology)
off_topology = Topology.from_openmm(openmm_topology=ligand_pdbfile.topology
,unique_molecules=[ligand])
# Load the smirnoff99Frosst system from disk.
force_field = ForceField('test_forcefields/smirnoff99Frosst.offxml')
# Parametrize the ligand molecule.
ligand_system = force_field.create_openmm_system(off_topology)
I have tried loading molecule from sdf and SMILES, but both return the following error:
ValueError Traceback (most recent call last)
<ipython-input-9-584e58c71b83> in <module>
6 print(ligand_pdbfile.topology)
7 off_topology = Topology.from_openmm(openmm_topology=ligand_pdbfile.topology
----> 8 ,unique_molecules=[ligand])
9
10 # Load the smirnoff99Frosst system from disk.
~/packages/miniconda3/envs/md/lib/python3.6/site-packages/openforcefield/topology/topology.py in from_openmm(cls, openmm_topology, unique_molecules)`
1661 if not (match_found):
1662 hill_formula = Topology._networkx_to_hill_formula(omm_mol_G)
-> 1663 raise ValueError(f'No match found for molecule {hill_formula}')
1664
1665 # The connected_component_subgraph function above may have scrambled the molecule order, so sort molecules
ValueError: No match found for molecule C1
I am using the following environment on an Ubuntu 16.04 machine:
openforcefield 0.4.0 py36_0 omnia
openmm 7.3.1 py36_cuda92_rc_2 omnia/label/cuda92
rdkit 2019.03.2 py36h9c20d5c_0 conda-forge
Is this an issue with my environment or am I messing up the script somehow?
 yabmtm
yabmtm
All 7 comments
Thanks for reporting this!
I'd like to capture a bit more information so we can reproduce this locally:
Can you share the PDB file (test.pdb) and the SDF file (test.sdf) you are loading? You can upload a ZIP archive containing these.
The No match found for molecule C1 error indicates that there is a discrepancy between the Molecule you are constructing and the molecule present in the PDB file. The most likely cause is that your PDB file does not follow the PDB format specification and is missing CONECT records that contain vital information about which atoms are connected to each other, so the toolkit thinks that you have an individual carbon atom (hill formula C1) instead of an actual molecule present in the PDB file.
There may be a simpler route than requiring a PDB file if you just want to simulate toluene. We're happy to give you pointers on the easiest way to do something if you can give us more information about your use case!
 jchodera
on 14 Jun 2019
jchodera
on 14 Jun 2019
@j-wags: I wonder if we can provide a more useful error message in cases like this. Perhaps there's an easy way to detect when the OpenMM Topology is contains single atoms that are not generally found as individual atoms, and to call this helpful Topology inspector when we fail to find a match for our molecule?
jchodera
on 14 Jun 2019
Agreed. I suspect that missing CONECT records are causing @yabmtm's problem (though it would be great to verify, if they could post their PDB). I've encountered this message before in that situation. I've opened an issue to add a more informative error.
 j-wags
on 14 Jun 2019
j-wags
on 14 Jun 2019
Here is an attachment containing relevant structure files. TAY is the molecule I am especially interested in modeling, test was just to see if it could work with a simpler structure.
pdb_files.zip
It looks like that's an issue. Neither of my pdb's contain CONECT records. I'm attaching a zip file with some files I attempted using. For the TAY molecule, I added CONECT records via Chimera, and tried again but am now receiving the same error except with
ValueError: No match found for molecule C15H19N5O1
Thanks for your timely responses.
My end goal is to use this workflow to parameterize small molecules for protein-ligand simulations in OpenMM. Our current workflow uses several different scripts to assign charges and GAFF atomtypes, and often returns missing term errors for bonding and dihedral terms. So after much frustration, I'm turning to Smirnoff to compare results from atomtyped topologies to ones generated here.
yabmtm
on 14 Jun 2019
@yabmtm : Thanks for including the SDF and PDB files!
It looks like your SDF file and PDB file described different molecules! The SDF file has two nitrogens protonated, while the PDB file is missing those protons. As a result, the OFF toolkit can't find an exact match for the molecular definition you specified in the SDF file within your PDB file.
Here's the SDF:
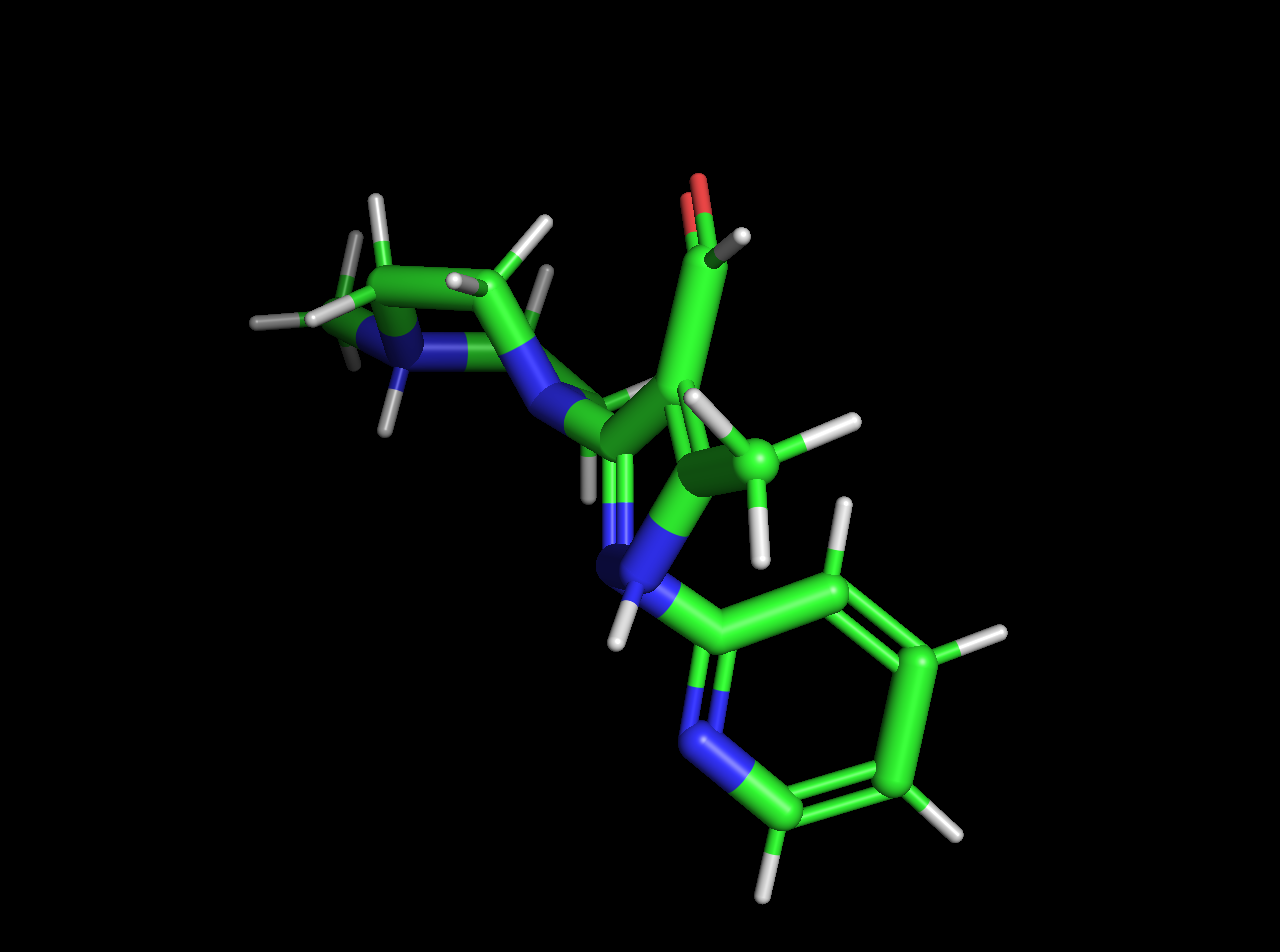
and here's the PDB:
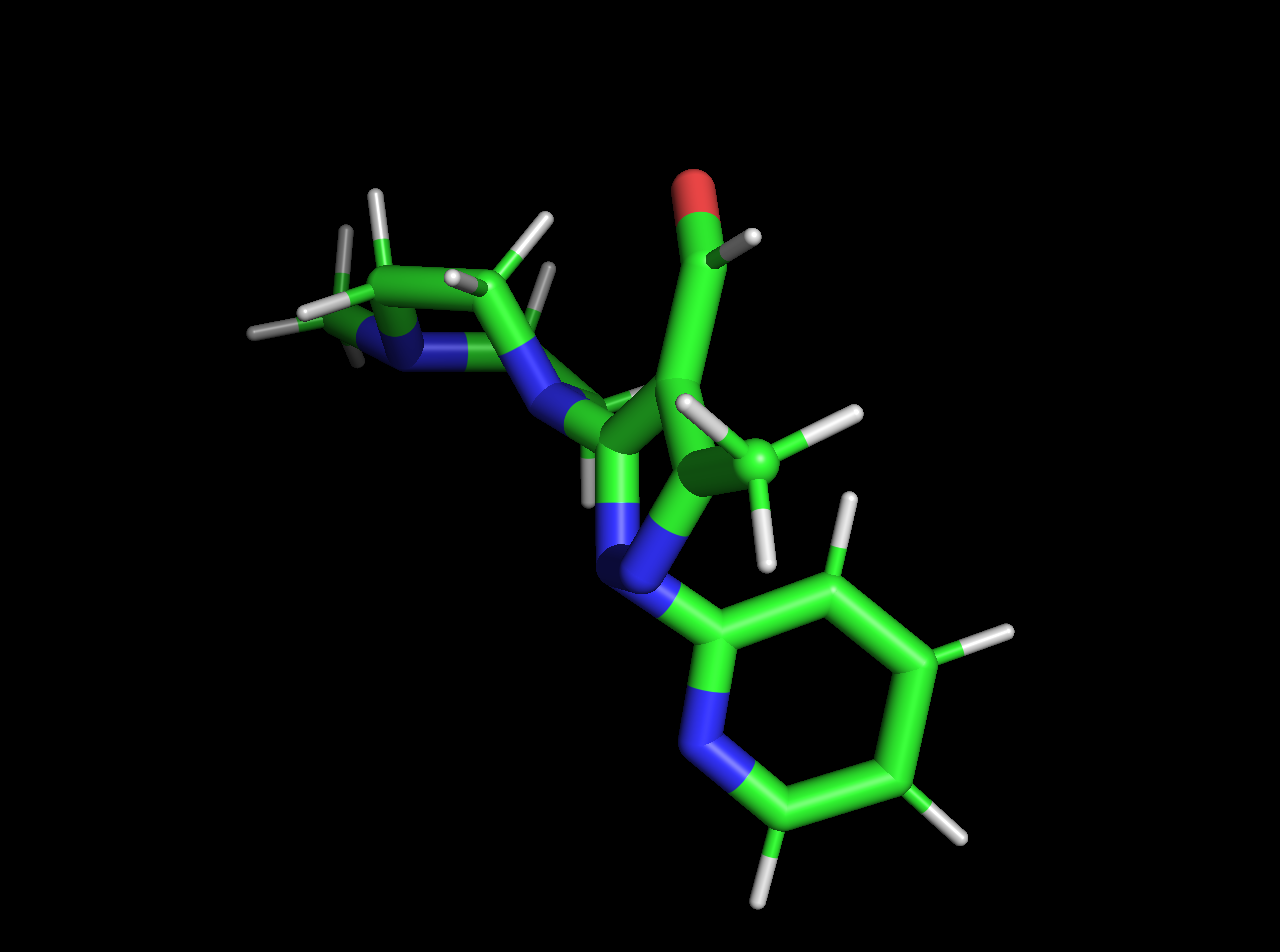
If you're just interested in simulating the molecule in the SDF file (which contains all the chemical information we need), you shouldn't need the PDB file as well---you should be able to just create a System from the Molecule you create from
from openforcefield.topology import Molecule, Topology
from openforcefield.typing.engines.smirnoff import ForceField
# Create an OpenFF Molecule
molecule = Molecule.from_file('test.sdf')
# Create an OpenFF Topology from that molecule
off_topology = molecule.to_topology()
# Load the smirnoff99Frosst system from disk.
force_field = ForceField('test_forcefields/smirnoff99Frosst.offxml')
# Parametrize the ligand molecule.
ligand_system = force_field.create_openmm_system(off_topology)
The PDB file handling code is just there in case you have a complex molecular system where you need to match specific molecules within the PDB file. We'll make much more extensive use of this later once OpenFF can apply both protein and small molecule parameters.
jchodera
on 16 Jun 2019
@j-wags : We could probably improve our error message in this case if we found a match for the heavy atom Hill formula, but the number of hydrogens was different. Something like "Your system contains a molecule with the same number of heavy atoms (C15N5O1), but the number of hydrogens is different. You might check all protons have been explicitly added in the desired protonation and tautomeric state."
jchodera
on 16 Jun 2019
Well, that's embarrassing. Thanks very much for your help. I just got it working and will let you know if I run into any other issues creating systems over the coming weeks. I'll look forward to keeping up with OpenFF and hopefully even contributing to it in the future!
yabmtm
on 16 Jun 2019
Related issues
 trevorgokey
·
4Comments
trevorgokey
·
4Comments
 nividic
·
5Comments
nividic
·
5Comments
 andrrizzi
·
7Comments
j-wags
·
4Comments
j-wags
·
3Comments
andrrizzi
·
7Comments
j-wags
·
4Comments
j-wags
·
3Comments
Most helpful comment
Well, that's embarrassing. Thanks very much for your help. I just got it working and will let you know if I run into any other issues creating systems over the coming weeks. I'll look forward to keeping up with OpenFF and hopefully even contributing to it in the future!