Openff-toolkit: Accept only explicit H SMILES?
Inspired by @ChayaSt's tracked issue at https://github.com/openforcefield/cmiles/issues/12
(I'm going to come back to this and edit in more details when I have time)
Briefly: Sometimes, when given a SMILES with implicit H's, a toolkit may have a choice of where to place an H. It's possible that OE and RDK will make this choice differently, yielding different molecules from the same implicit H SMILES. We rely on these toolkits' explicit-hydrogen-addition functions, so this could be a problem.
Current stage: I haven't looked too far into this, but it appears that might occur.
insert example of a case of a valid SMILES where OE and RDK place H's differently here
 j-wags
j-wags
All 8 comments
For now, I suggest we try to be permissive in the inputs we accept from users via the public API, since being less permissive is likely to frustrate users who just want to create a molecule.
We should definitely note this potential toolkit dependence in the documentation, however, and recommend explicit hydrogen SMILES be used if hydrogen tautomer selection is critical.
Internally, we can be sure to use explicit-hydrogen SMILES in any representations to avoid the potential for accidental transmogrifications.
In future, we can add a strict flag that users who want to be sure that toolkit interoperability is guaranteed can use, and then we can raise exceptions if we detect anything is ambiguously specified in a way that may produce toolkit-dependent results.
 jchodera
on 6 Dec 2018
jchodera
on 6 Dec 2018
Definitely agree on the strict flag.
We should definitely note this potential toolkit dependence in the documentation, however, and recommend explicit hydrogen SMILES be used if hydrogen tautomer selection is critical.
Adding to the documentation. Should go in the next commit.
Internally, we can be sure to use explicit-hydrogen SMILES in any representations to avoid the potential for accidental transmogrifications.
Agreed.
For now, I suggest we try to be permissive in the inputs we accept from users via the public API, since being less permissive is likely to frustrate users who just want to create a molecule.
Ok. This is along the lines of what I was thinking. This may make lead to a few misunderstandings/undesired molecule interpretations, but I think the benefits of being permissive outweigh the cons at this time.
j-wags
on 6 Dec 2018
Being permissive and warning is a good plan.
We bridge two vastly different worlds. Academics basically want to feed "whatever" in and have it work, and pharma people want to be able to feed "this specific molecule and tautomer I have already carefully prepared and don't want anyone messing with" and have it behave. The former group needs to be warned when they do something ill advised, and eventually also have help cleaning up and checking their chemistry (there is an issue on this somewhere) but the latter group (who is funding this at present) just needs it to work with their carefully prepared molecules.
 davidlmobley
on 8 Dec 2018
davidlmobley
on 8 Dec 2018
So, as confirmation that this the same implicit-H SMILES could be interpreted differently:
@ChayaSt found an example of a not-super-exotic molecule that gets a different number of explicit H's assigned by OE vs. RDKit. We'll need to dive into the Daylight SMILES spec to figure out if there's a single clear way to interpret the SMILES (and one of the toolkits is wrong), or if this is a grey area.
Copying her Slack post here:
```sm = 'c1ccc2c(c1)cc(N2)O'
mol = oechem.OEMol()
oechem.OESmilesToMol(mol, sm)
print(sum([atom.GetImplicitHCount() for atom in mol.GetAtoms()]))
oechem.OEAddExplicitHydrogens(mol)
new_sm_oe = oechem.OECreateSmiString(mol, oechem.OESMILESFlag_Hydrogens | oechem.OESMILESFlag_ISOMERIC)
mol_rd = Chem.MolFromSmiles(sm)
print(sum([atom.GetTotalNumHs(False) for atom in mol_rd.GetAtoms()]))
with explicit H SMILES
mol_rd = Chem.MolFromSmiles(new_sm_oe)
print(sum([atom.GetTotalNumHs(False) for atom in mol_rd.GetAtoms()]))
output:
Warning: OEParseSmiles() : unable to Kekulize SMILES:
Warning: c1ccc2c(c1)cc(N2)O
8
7
8
```
OEMol:
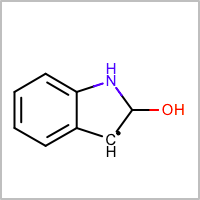
It interprets this SMILES as a carbon radical
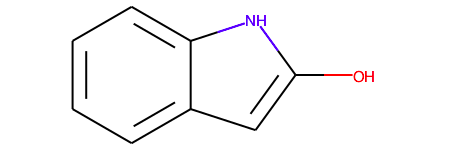
RDKit interprets it as an aromatic carbon.
RDMol:
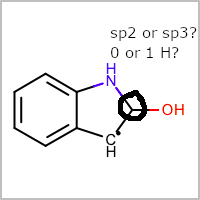
Annotated RDMol:
j-wags
on 10 Dec 2018
Is that a well-formed SMILES? The capital N means that the nitrogen is not aromatic, while the RDKit version of the structure has the nitrogen in an aromatic ring (which would be n).
My guess is that we want to do some pre-cleaning of our small molecule datasets by running them through the toolkits and making sure that both toolkits agree on what the molecule is. We can reject any discrepancies into a pile for later curation.
jchodera
on 10 Dec 2018
Is that a well-formed SMILES? The capital N means that the nitrogen is not aromatic, while the RDKit version of the structure has the nitrogen in an aromatic ring (which would be n).
No, it is not a well-formed SMILES and that was partially the point of this example. OpenEye raises a warning for this but RDKit just reads it as aromatic. RDKit will interpret it the same as OpenEye if the SMILES has explicit H.
My guess is that we want to do some pre-cleaning of our small molecule datasets by running them through the toolkits and making sure that both toolkits agree on what the molecule is. We can reject any discrepancies into a pile for later curation.
Yes. This will prevent problematic inputs as the one above. Requiring explicit H SMILES can prevent garbage input like this but your suggestion is better because it allows permissive inputs. However, this assumes that users will have OpenEye license.
 ChayaSt
on 10 Dec 2018
ChayaSt
on 10 Dec 2018
We certainly can't assume that pharma users will have an OpenEye license, but we can assume that we (the Open Force Field Initiative) will have an OpenEye license and will want to both pre-filter our molecule datasets using both toolkits to drop problematic molecules and operate the ToolkitWrapper in a "check to make sure both toolkits agree" mode during production to catch problems.
jchodera
on 10 Dec 2018
Agree with John that we want to make sure our molecules are clean before doing anything with them and it's appropriate to use both toolkits to check before we do anything with them.
Ensuring users' molecules are clean is a harder problem and should be dealt with separately.
davidlmobley
on 18 Dec 2018
Related issues
j-wags
·
7Comments
j-wags
·
4Comments
j-wags
·
3Comments
 jthorton
·
4Comments
jthorton
·
4Comments
 maxentile
·
5Comments
maxentile
·
5Comments
Most helpful comment
For now, I suggest we try to be permissive in the inputs we accept from users via the public API, since being less permissive is likely to frustrate users who just want to create a molecule.
We should definitely note this potential toolkit dependence in the documentation, however, and recommend explicit hydrogen SMILES be used if hydrogen tautomer selection is critical.
Internally, we can be sure to use explicit-hydrogen SMILES in any representations to avoid the potential for accidental transmogrifications.
In future, we can add a
strictflag that users who want to be sure that toolkit interoperability is guaranteed can use, and then we can raise exceptions if we detect anything is ambiguously specified in a way that may produce toolkit-dependent results.